






With thanks to Amelia Meier-Batschelet, Johanna Huggler, and Martin Meyer for help with compilation of this article.
 For the podcast associated with this article, please visit https://dbpia.nl.go.kr/eurheartj/pages/Podcasts.
For the podcast associated with this article, please visit https://dbpia.nl.go.kr/eurheartj/pages/Podcasts.
This issue opens with the Special Article ‘Twitter promotion is associated with higher citation rates of cardiovascular articles: the ESC Journals Randomized Study’.1 In this contribution, Ricardo Ladeiras-Lopes from the University of Porto in Portugal and colleagues (myself among them) indicate that the association between the dissemination of scientific articles on Twitter and online visibility (as assessed by the Altmetric Score) is still controversial, and the impact on citation rates has never been rigorously addressed for cardiovascular medicine journals using a randomized design. The ESC Journals Study randomized 695 papers published in the ESC Journal Family (March 2018–May 2019) for promotion on Twitter or to a control arm (with no active tweeting from ESC channels) and aimed to assess whether Twitter promotion was associated with an increase in citation rates (primary endpoint) and of the Altmetric Score. This is the final analysis including 694 articles (one paper excluded due to retraction). After a median follow-up of 994 days, Twitter promotion of articles was associated with a 1.12 [95% confidence interval (CI) 1.08–1.15] higher rate of citations, and this effect was independent of the type of article. The Altmetric Score and number of users tweeting were positive predictors for the number of citations. Thus, a social media strategy of Twitter promotion for cardiovascular medicine papers seems to be associated with increased online visibility and higher numbers of citations.
This issue has a focus on vascular biology and medicine. Given the role of low density lipoproteins (LDLs) as a principal driving force in the development of atherosclerosis, pharmaceutical interventions so far largely focused on LDL-lowering strategies. However, despite substantial progress in LDL lowering and the control of other risk factors, residual rates of cardiovascular disease remain high, calling for the identification of novel treatment paradigms for atherosclerosis.2–4 In a State of the Art Review article entitled ‘Targeting the CCL2–CCR2 axis for atheroprotection’, Marios K. Georgakis from the Harvard Medical School in Boston, MA, USA, and colleagues note that decades of research have established atherosclerosis as an inflammatory disease.5 Only recently, however, clinical trials provided proof-of-concept evidence for the efficacy of anti-inflammatory strategies with respect to cardiovascular events, thus offering a new paradigm for lowering residual vascular risk. Efforts to target the inflammasome–interleukin-1β–interleukin-6 pathway have been highly successful, but interindividual variations in drug response, a lack of reduction in all-cause mortality, and a higher rate of infections also highlight the need for a second generation of anti-inflammatory agents targeting atherosclerosis-specific immune mechanisms while minimizing systemic side effects. CC-motif chemokine ligand 2/monocyte-chemoattractant protein-1 (CCL2/MCP-1) orchestrates inflammatory monocyte trafficking between the bone marrow, circulation, and atherosclerotic plaques by binding to its cognate receptor C-C chemokine receptor type 2 (CCR2). Adding to a strong body of data from experimental atherosclerosis models, a coherent series of recent large-scale genetic and observational epidemiological studies along with data from human atherosclerotic plaques highlight the relevance and therapeutic potential of the CCL2–CCR2 axis in human atherosclerosis. Here, the authors summarize experimental and human data pinpointing the CCL2–CCR2 pathway as an emerging drug target in cardiovascular disease. Furthermore, they contextualize previous efforts to interfere with this pathway, scrutinize approaches of ligand targeting vs. receptor targeting, and discuss possible pathway-intrinsic opportunities and challenges related to pharmacological targeting of the CCL2–CCR2 axis in human atherosclerotic disease.
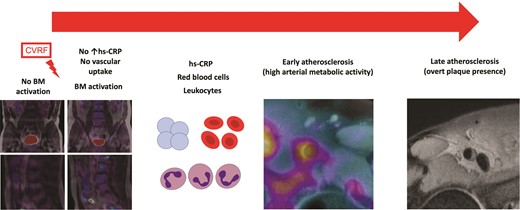
The hypothesis of the natural history of the inflammatory process involving atherosclerotic plaque formation. Bone marrow (BM) is implicated in the atherosclerotic process long before the appearance of acute cardiovascular events. Cardiovascular risk factors trigger BM activation, initially in the absence of systemic inflammation. As BM activation progresses, it is accompanied by an increase in haematopoietic progenitor cells and an associated increase in inflammatory markers. The next step in the process is arterial inflammation, leading to an increase in atherosclerotic burden.8 CVRF, cardiovascular risk factors; hs-CRP, high sensitivity C-reactive protein.
Experimental studies suggest that increased bone marrow activity is involved in the association between cardiovascular risk factors and inflammation in atherosclerosis.6,7 However, human data to support this association are sparse. In a Clinical Research article entitled ‘Bone marrow activation in response to metabolic syndrome and early atherosclerosis’, Ana Devesa from the Icahn School of Medicine at Mount Sinai in New York, NY, USA, and colleagues studied the association between cardiovascular risk factors, bone marrow activation, and subclinical atherosclerosis.8 Whole body vascular [18F]fluorodeoxyglucose positron emission tomography/magnetic resonance imaging (18F-FDG PET/MRI) was performed in 745 apparently healthy individuals (median age 50 years, 84% men) from the Progression of Early Subclinical Atherosclerosis (PESA) study. Bone marrow activation (defined as bone marrow [18F]FDG uptake above the median maximal standardized uptake value) was assessed in the lumbar vertebrae (L3–L4). Systemic inflammation was indexed from circulating biomarkers. Early atherosclerosis was evaluated by arterial metabolic activity by [18F]FDG uptake in five vascular territories. Late atherosclerosis was evaluated by fully formed plaques on MRI. Subjects with bone marrow activation more frequently had metabolic syndrome (MetS) (22.2 vs. 6.7%, P < 0.001). Bone marrow activation was significantly associated with all MetS components. Bone marrow activation was also associated with increased haematopoiesis—characterized by significantly elevated leucocyte (mainly neutrophil and monocytes) and erythrocyte counts—and with markers of systemic inflammation including high-sensitivity C-reactive protein, ferritin, fibrinogen, P-selectin, and vascular cell adhesion molecule-1. The associations between bone marrow activation and MetS (and its components) and increased erythropoiesis were maintained in the subgroup of participants with no systemic inflammation. Bone marrow activation was significantly associated with high arterial metabolic activity ([18F]FDG uptake). The co-occurrence of bone marrow activation and arterial [18F]FDG uptake was associated with more advanced atherosclerosis (i.e. plaque presence and burden) (Figure 2).
Devesa et al. conclude that bone marrow activation is associated with early atherosclerosis, characterized by high arterial metabolic activity. Bone marrow activation appears to be an early phenomenon in atherosclerosis development. The contribution is accompanied by an Editorial by Peter Libby and Matthias Nahrendorf from Harvard Medical School in Boston, MA, USA, and Filip K. Swirski from the Icahn School of Medicine at Mount Sinai in New York, NY, USA.9 Libby and colleagues conclude that overall, this important study provides key translational support for the burgeoning findings in mice that implicate altered bone marrow function and enhanced haematopoiesis as a previously unrecognized driver of atherogenesis in humans. This new work from PESA validates concepts that have emerged from recent laboratory experiments. The production of inflammatory leucocytes in haematopoietic organs has emerged as a newly recognized mechanism promoting cardiovascular diseases. This ‘primary prevention’ study in PESA provides us with novel information gleaned from assessment of a marker of bone marrow activation in humans. We have a new therapeutic target on the horizon in cardiovascular prevention: mitigating mischief in the bone marrow.
Hypogonadism is associated with cardiovascular disease. However, the cardiovascular impact of hypogonadism during development is unknown. In a Clinical Research article entitled ‘Vascular dysfunction and increased cardiovascular risk in hypospadias’, Angela K. Lucas-Herald from the University of Glasgow in the UK, and colleagues note that using hypospadias as a surrogate of hypogonadism, they investigated whether hypospadias is associated with vascular dysfunction and is a risk factor for cardiovascular disease.10 This human study spanned molecular mechanistic to epidemiological investigations. Clinical vascular phenotyping was performed in adolescents with hypospadias and controls. Small subcutaneous arteries from penile skin from boys undergoing hypospadias repair and controls were isolated and functional studies were assessed by myography. Vascular smooth muscle cells were used to assess: Rho kinase, reactive oxygen species (ROS), nitric oxide synthase/nitric oxide, and DNA damage. Systemic oxidative stress was assessed in plasma and urine. In adolescents with hypospadias, systolic blood pressure (P = 0.005), pulse pressure (P = 0.03), and carotid intima-media thickness standard deviation scores (P = 0.01) were increased as compared with controls. Arteries from boys with hypospadias demonstrated increased U46619-induced vasoconstriction (P = 0.009) and reduced acetylcholine-induced endothelium-dependent (P < 0.0001) and sodium nitroprusside-induced endothelium-independent vasorelaxation (P < 0.0001). Men born with hypospadias were at increased risk of arrhythmias [odds ratio (OR) 2.8, P = 0.003], hypertension (OR 4.2, P = 0.04), and heart failure (OR 1.9, P = 0.02). Underlying mechanisms involved perturbed Rho kinase- and Nox5/ROS-dependent signalling.
Lucas-Herald et al. conclude that hypospadias is associated with vascular dysfunction and predisposes to hypertension and cardiovascular disease in adulthood. These novel findings delineate molecular mechanisms of vascular injury in hypogonadism and identify hypospadias as a cardiovascular risk factor in males. The contribution is accompanied by an Editorial by Nicolle Kränkel from the Charité–Universitätsmedizin Berlin in Germany.11 Kränkel concludes that the study by Lucas-Herald et al. stresses the intricate interactions between sex hormone action at different developmental stages and lifetime cardiovascular risk. Even after surgical ‘solution’ of the ‘cosmetic problem’, the (postulated) presence of predisposing molecular patterns and underlying or confounding factors requires our attention in view of improved cardiovascular risk screening and management.
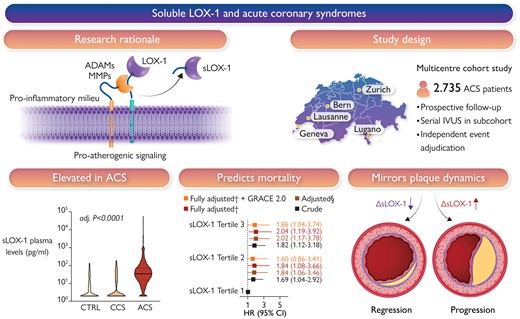
Atherosclerotic plaques express high levels of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), with plaque regions particularly prone to instability showing accentuated LOX-1 abundance. The proinflammatory milieu within atherosclerotic plaques enhances LOX-1 synthesis, promotes LOX-1 shedding, and thus soluble LOX-1 (sLOX-1) release. The turnover of membrane-bound LOX-1 determines plaque composition and thus stability, with systemically circulating sLOX-1 emerging as a novel biomarker reflecting plaque burden and vulnerability. In this multicentre prospective cohort study, it was found that plasma levels of sLOX-1 are markedly elevated in patients presenting with acute coronary syndromes (ACS) when compared with both chronic coronary syndrome (CCS) and healthy subjects (CTRL). High sLOX-1 levels were independently associated with a higher multivariable-adjusted 1-year mortality risk following ACS, a finding that remained consistent after controlling for GRACE 2.0. In patients subjected to serial intravascular ultrasound following the index ACS, sLOX-1 dropped markedly in those with coronary plaque regression, while persistently high sLOX-1 levels were associated with plaque progression. §Adjusted for sex, age, ACS type, hs-CRP, history of hypercholesterolaemia, eGFR, LDL-C, and diagnosis of diabetes; †adjusted for sex, age, ACS type, hs-CRP, history of hypercholesterolaemia, eGFR, LDL-C, diagnosis of diabetes, NT-proBNP, and hs-TnT. ADAM, a disintegrin and metalloproteinase; GRACE, Global Registry of Acute Coronary Events; IVUS, intravascular ultrasound; MMP, matrix metalloproteinase; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; sLOX-1, soluble LOX-1.21
The dynamics of plaque evolution and stability are shaped by an interplay of factors that promote or mitigate atherogenesis.12–20 Pre-clinical evidence implicates lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in key steps of the disease process. Indeed, initially described as the main scavenger receptor for endothelial oxidized LDL uptake, LOX-1 is increasingly acknowledged as a key factor determining plaque progression and stability. LOX-1 and its shedding product [soluble LOX-1 (sLOX-1)] are implicated in atherosclerotic cardiovascular disease (ASCVD) pathogenesis. In another Clinical Research article entitled ‘Soluble lectin-like oxidized low-density lipoprotein receptor-1 predicts premature death in acute coronary syndromes’, Simon Kraler from the University of Zurich in Switzerland, and colleagues examined the relationship of sLOX-1 to both fatal events and plaque progression in patients with acute coronary syndromes (ACS).21 Plasma sLOX-1 was assessed at baseline in ACS and chronic coronary syndrome (CCS) patients prospectively recruited in the multicentre SPUM-ACS study, with sex- and age-matched healthy subjects serving as additional controls (n = 2924). Compared with both CCS and controls, ACS patients showed markedly elevated sLOX-1 levels (median, 2.00 and 2.00 vs. 35.08 pg/mL; P < 0.0001) which were independently associated with increased mortality risk over 30 days [top vs. bottom tertile: adjusted hazard ratio (HR) 3.11; P = 0.0055] and at 1 year (adjusted HR 2.04; P = 0.0098). Results remained consistent after adjustment for GRACE 2.0 (adjusted HR 1.86; P = 0.0391) and were primarily driven by the pronounced relationship of sLOX-1 with cardiovascular mortality at 30 days (adjusted HR, 3.81; P = 0.0036) and at 1 year (adjusted HR 2.29; P = 0.0148). In ACS patients undergoing serial intracoronary imaging and statin therapy, sLOX-1 dropped significantly in those with coronary plaque regression at 1 year and showed a good discrimination for predicting plaque progression (area under the curve = 0.74; P = 0.0031) (Figure 2).
Kraler and colleagues conclude that plasma sLOX-1 levels are increased during ACS and predict fatal events beyond traditional and emerging risk factors. Persistently high sLOX-1 associates with coronary plaque progression in patients with established ASCVD. The contribution is accompanied by an Editorial by Andrei C. Sposito from State University of Campinas (Unicamp) in Sao Paulo, Brazil.22 Sposito concludes that there is accumulating evidence indicating a strong relationship between sLOX-1 and the presence and complexity of coronary artery disease. Kraler et al. have demonstrated an even more relevant role for sLOX-1—the prediction of the risk of death after ACS in an additive way to risk algorithms and traditional biomarkers. Considering the findings of Kraler et al. and the above-mentioned mechanistic data, it is reasonable to infer the existence of multiple and complex interactions involving both the myocardium and the arterial wall. From where we stand now, sLOX-1 must be considered among the potent prognostic biomarkers for CVD, particularly during the acute inflammatory response such as in patients with ACS.
Macrophages perform both pro- and antiatherogenic functions depending on their programming.23,24 Rewiring macrophage transcriptional states is an attractive therapeutic strategy for ASCVD. The transcription factor interferon regulatory factor-5 (IRF5) is a master regulator of macrophage activation, important in murine atherogenesis. Its role in human atherosclerosis and its complications are unknown. IRF5 drives macrophages towards a proinflammatory state. In a Translational Research article entitled ‘Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques’, Andreas Edsfeldt from the Lund University in Malmö, Sweden, and colleagues investigated the role of IRF5 in human atherosclerosis and plaque stability.25 Bulk RNA sequencing from the Carotid Plaque Imaging Project biobank were used to mine associations between major macrophage-associated genes and transcription factors and human symptomatic carotid disease. Immunohistochemistry, proximity extension assays, and Helios cytometry by time of flight (CyTOF) were used for validation. The effect of IRF5 deficiency on carotid plaque phenotype and rupture in ApoE−/− mice was studied in an inducible model of plaque rupture. IRF5 and ITGAX/CD11c were identified as the macrophage-associated genes with the strongest associations with symptomatic carotid disease. Expression of IRF5 and ITGAX/CD11c correlated with the vulnerability index, proinflammatory plaque cytokine levels, necrotic core area, and with each other. Macrophages were the predominant CD11c-expressing immune cells in the plaque by CyTOF and immunohistochemistry. IRF5-immunopositive areas were predominantly found within CD11c+ areas, with a predilection for the shoulder region, the area of the human plaque most prone to rupture. Accordingly, an inducible plaque rupture model of ApoE−/−Irf5−/− mice had significantly lower frequencies of carotid plaque ruptures, smaller necrotic cores, and fewer CD11c+ macrophages than their IRF5-competent counterparts.
The authors conclude that using complementary evidence from data from human carotid endarterectomies and a murine model of inducible rupture of carotid artery plaque in IRF5-deficient mice, they demonstrate a mechanistic link between the proinflammatory transcription factor IRF5, macrophage phenotype, plaque inflammation, and its vulnerability to rupture. The manuscript is accompanied by an Editorial by Alexandra A.C. Newman, Yannick Cyr, and Kathryn J. Moore from the New York University Grossman School of Medicine in New York, NY, USA.26 The authors state that a notable aspect of the study by Edsfeldt et al. is the integration of multi-omic analyses from unstable human lesions to identify clinically relevant targets against the key inflammatory processes that drive plaque rupture and its sequelae. Many pathways of relevance to murine atherosclerosis have failed to be translated into viable therapeutic strategies for human atherosclerosis. Importantly, the majority of mouse models used to study atherosclerosis do not recapitulate plaque rupture seen in humans. In this contribution, the authors used a reverse translation strategy to mechanistically test the role of IRF5, identified as a key factor correlating with plaque vulnerability in humans, in a mouse model of inducible plaque rupture. Combined, these approaches establish that IRF5 maintains a relatively small subset of cells within human lesions (CD11c+ macrophages, <1% of the human carotid lesion area), contributes significantly to inflammation, and correlates with lesion instability. The rational targeting of pathways that contribute to pathological inflammation, such as IRF5, represents a powerful approach to reduce plaque rupture and associated adverse cardiac events without interfering with immune defence against pathogens.
The issue is also complemented by two Discussion Forum contributions. In a commentary entitled ‘Is there an increased incidence of subclinical proximal deep vein thrombosis after mild to moderate course of SARS-CoV-2 infection?’ Lars Müller from the Dermatologikum Hamburg in Germany, and colleagues comment on the recent publication ‘Multi-organ assessment in mainly non-hospitalized individuals after SARS-CoV-2 infection: The Hamburg City Health Study COVID programme’ by Elina Larissa Petersen from the University Heart and Vascular Center in Hamburg, Germany.27,28 The authors respond in a separate comment.29
Dr. Crea reports speaker fees from Amgen, Astra Zeneca, Servier, BMS, other from GlyCardial Diagnostics, outside the submitted work.
The editors hope that this issue of the European Heart Journal will be of interest to its readers.


{kind=link}
{kind=link}
{kind=link}