





Abstract
Mutation type, location, dominant-negative I Ks reduction, and possibly loss of cyclic adenosine monophosphate (cAMP)-dependent I Ks stimulation via protein kinase A (PKA) influence the clinical severity of long QT syndrome type 1 (LQT1). Given the malignancy of KCNQ1-p.A341V, we assessed whether mutations neighbouring p.A341V in the S6 channel segment could also increase arrhythmic risk.
Clinical and genetic data were obtained from 1316 LQT1 patients [450 families, 166 unique KCNQ1 mutations, including 277 p.A341V-positive subjects, 139 patients with p.A341-neighbouring mutations (91 missense, 48 non-missense), and 900 other LQT1 subjects]. A first cardiac event represented the primary endpoint. S6 segment missense variant characteristics, particularly cAMP stimulation responses, were analysed by cellular electrophysiology. p.A341-neighbouring mutation carriers had a QTc shorter than p.A341V carriers (477 ± 33 vs. 490 ± 44 ms) but longer than the remaining LQT1 patient population (467 ± 41 ms) (P < 0.05 for both). Similarly, the frequency of symptomatic subjects in the p.A341-neighbouring subgroup was intermediate between the other two groups (43% vs. 73% vs. 20%; P < 0.001). These differences in clinical severity can be explained, for p.A341V vs. p.A341-neighbouring mutations, by the p.A341V-specific impairment of I Ks regulation. The differences between the p.A341-neighbouring subgroup and the rest of LQT1 mutations may be explained by the functional importance of the S6 segment for channel activation.
KCNQ1 S6 segment mutations surrounding p.A341 increase arrhythmic risk. p.A341V-specific loss of PKA-dependent I Ks enhancement correlates with its phenotypic severity. Cellular studies providing further insights into I Ks-channel regulation and knowledge of structure-function relationships could improve risk stratification. These findings impact on clinical management.
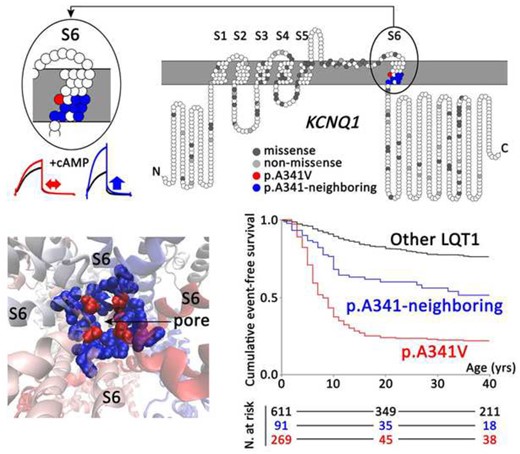
Upper panel: Topology of KCNQ1 missense and non-missense mutations identified in all studied long QT syndrome type 1 patients (n = 1316), according to their amino acid position along the KCNQ1 channel. Location of p.A341V and p.A341-neighbouring mutations in the KCNQ1-S6 segment is highlighted in red and blue, respectively. Exemplary I Ks traces for p.A341V (red) and p.A341V-neighbouring mutation (blue), with and without cyclic adenosine monophosphate/okadaic acid are depicted on the left. Left lower panel: structural image of the pore of the I Ks channel. Amino acid position A341 is indicated in red and the A341-neighbouring amino acids in blue. Right lower panel: Cumulative event-free survival of patients with p.A341V (red; n = 269), p.A341-neighbouring mutations (blue; n = 91), and all other KCNQ1-missense mutations located outside the S6 region (black; n = 611).
See page 4756 for the editorial comment for this article ‘Does knowledge of the mutation in hereditary long QT syndrome aid risk stratification?’, by A. Tinker, https://doi.org/10.1093/eurheartj/ehab668.
Introduction
The congenital long QT syndrome (LQTS), with a prevalence of ∼1 in 2000,1 is the best understood cardiogenetic disorder, often causing life-threatening arrhythmias.2 Progress made in the last 50 years has been mind-boggling,3–5 and unravelling of the genetic bases6 has strengthened the concept that LQTS is an insightful paradigm for sudden cardiac death (SCD). Of the three major genetic subgroups, the most prevalent is LQTS type 1 (LQT1), caused by loss-of-function mutations in the KCNQ1 gene,7 encoding the alpha-subunit of the cardiac Kv7.1 potassium channel, conducting the slowly activating delayed-rectifier K+ current I Ks. 8 I Ks is regulated by the sympathetic nervous system, which exerts its control on heart rate through β-adrenergic receptor activation. Under normal conditions, β-adrenergic receptor activation (as part of sympathetic stimulation) modulates channel function through phosphorylation that enhances I Ks and thereby contributes significantly to action-potential shortening.9 This physiological response specifically requires cyclic adenosine monophosphate (cAMP)-dependent activation of protein kinase A (PKA) to phosphorylate KCNQ1, a mechanism that also involves the A-kinase anchoring protein 9, also known as yotiao, an adaptor protein that presents PKA to the I Ks-channel complex.10 Phosphorylation also requires the co-assembly of KCNQ1 with KCNE1, its auxiliary subunit.11 The impairment of I Ks prevents proper shortening of the QT interval during sympathetically driven heart rate increases and this may trigger lethal arrhythmias during exercise, emotional stress, and swimming.12 KCNQ1 mutations that cripple basal I Ks and undermine β-adrenergic I Ks stimulation facilitate repolarization prolongation by the overriding of inward currents, with exaggerated regional dispersion of repolarization and premature ventricular beats as culminating factors that precipitate torsades-de-pointes ventricular tachycardia. These mechanisms link the genetic and biophysical characteristics of KCNQ1 mutations with sympathetically driven tachyarrhythmias in susceptible patients.
It has become evident that mutation type (missense vs. non-missense, i.e. nonsense, frameshift, in-frame insertions/deletions and splice-site mutations) and location (transmembrane and pore region, cytoplasmic loop vs. N- or C-terminus13–15) are differently associated with clinical severity but, in general, how and why remains poorly understood. Similarly, dominant-negative mutations appear to be more severe than those causing haploinsufficiency.14 We have previously identified the hotspot KCNQ1-p.A341V mutation as particularly malignant, both in a South African LQTS founder population16 , 17 and in worldwide patient cohorts from different ethnic backgrounds with this mutation.18 Recent reports have also highlighted the importance of loss of PKA-dependent I Ks regulation conferred by the KCNQ1 S6 segment pathogenic variant p.A341V19 and several cytoplasmic loop mutations.15 In these, the significant reduction of basal I Ks is further compromised by the loss of PKA-dependent I Ks enhancement, contributing to a severe clinical phenotype.15 , 19 Conversely, KCNQ1 mutations causing either (i) dominant-negative I Ks suppression, but preserved enhancement,20 or (ii) only mild basal I Ks reduction by haploinsufficiency but PKA insensitivity,21 may underlie LQT1 with a lower probability of arrhythmic events.
On this background, we compared the clinical characteristics of 1316 LQT1 patients, with 166 unique KCNQ1 mutations. We specifically focused on p.A341V and the p.A341-neighbouring mutations in the S6 segment of the channel protein and correlated clinical severity with cellular electrophysiology data in the absence or presence of cAMP to stimulate mutant I Ks. We tested the hypothesis that, similarly to p.A341V, p.A341V-neighbouring mutations could also confer a significant aggravation of the clinical phenotype and, if so, whether this could be attributed to a concomitant reduction of basal I Ks and loss of PKA-dependent regulation, as per p.A341V.
Methods
This study conforms to the Declaration of Helsinki and was performed according to a protocol approved by the local ethics committees of the five medical centres involved. Patients gave written informed consent for DNA diagnostics, electrocardiogram (ECG), and cellular research and clinical follow-up.
Clinical characteristics of the study population
Data sources and collection
The study population was composed through a collaborative project involving five centres from Italy, Norway, France, Russia, and South Africa. Data were obtained from 1316 LQT1 patients: 277 p.A341V-positive subjects, in part previously reported16 , 18 and including 195 individuals from a large South African LQTS founder population and 82 worldwide patients with different ethnic backgrounds, 139 patients with p.A341-neighbouring mutations, and 900 carriers of KCNQ1 mutations in areas other than the S6 segment. Jervell–Lange-Nielsen syndrome patients22 were not included. In all subjects, genetic and clinical data were obtained, including mutation details, demographic information, personal and family history of disease, type and timing of symptoms, ECG findings both off and on β-blockers, other treatment, and response to therapy. The QT interval was measured on the first ECG available, as always done in the studies from the International Registry for LQTS;14 these measurements were made on ECGs at rest; we did not use the ECGs during exercise because the much faster heart rates, combined with the inherent measurement errors, would have reduced the reliability of our data. The QT interval was corrected for heart rate (QTc) according to Bazett’s formula,23 valid also for neonates.24 For simplicity, and given the variable penetrance of individual genetic variants, throughout the text, we interchangeably use the expressions ‘mutation carrier’ and ‘mutation positive subject’.
Endpoint
The primary endpoint was a first cardiac event occurring from birth through age 40 years. Cardiac events were defined as syncope (fainting spells with transient, but complete, loss of consciousness), aborted cardiac arrest (ACA) requiring resuscitation and LQTS-related SCD. Sudden cardiac deaths in family members judged to be LQTS-related according to established criteria25 were assumed to have occurred in genotype-positive subjects and included, even in the absence of genotyping and ECG documentation. Appropriate shocks from an implanted cardioverter-defibrillator (ICD) were considered as major arrhythmic events. Based on the history of cardiac events, mutation carriers were classified as symptomatic or asymptomatic. Symptomatic subjects had experienced at least one cardiac event irrespective of therapy and of age at onset. Asymptomatic mutation carriers were further distinguished into (i) ‘possibly’ asymptomatic, if they were younger than 18 years and had no events in the absence of β-blockers or if they had no events and initiated β-blockers before age 18 and (ii) ‘truly’ asymptomatic, if they were older than age 18 and without any previous cardiac events off therapy. This categorization, already established,16 allows for a more stringent definition of asymptomatic status while considering the natural risk exposure by age and the protective effect of β-blocker therapy. In addition to the occurrence of any first cardiac event, and consistent with our previous reports, we considered as markers of clinical severity a QTc >500 ms and the prevalence of major arrhythmic events (ACA/appropriate ICD shocks/SCD). We determined the efficacy of β-blockers by quantifying the occurrence of events while on therapy in those mutation carriers with precise information on treatment and outcome, with at least 1 year of follow-up after the initiation of therapy and for those who started β-blocker therapy before age 41 to avoid the confounding role of possible comorbidities. With the exception of known long-standing withdrawals, all the events occurring during sporadic omission of treatment were considered.
Genetic testing
Genetic testing was conducted with standard genetic protocols through either next-generation sequencing gene panels or Sanger sequencing. All clinically relevant genetic variants identified were confirmed with Sanger sequencing and were classified in accordance with the American College of Medical Genetics and Genomics and the Association of Molecular Pathology (ACMG) guidelines.26 Genetic variation was interrogated across different population databases, mainly gnomAD, but also the 1000 Genomes project browser, ClinVar, and the Human Gene Mutation Database, among others.
Subjects carrying multiple mutations were not included. KCNQ1 clinically relevant variants were also categorized according to location, with topological domains assigned according to the UniProt knowledgebase27 (UniProtKB—P51787, KCNQ1_HUMAN), and type, i.e. being missense or non-missense (splice site, in-frame insertions/deletions, nonsense, and frameshift). We evaluated the arrhythmic risk associated within SEVEN main genetic subgroups:
missense p.A341V (S6, residue 341);
missense p.A341-neighbouring mutations, i.e. mutations in the S6 segment (S6, residues 328–348), other than p.A341V;
missense mutations in all transmembrane segments other than S6, including the extracellular loops (S1–S5, residues 122–168, 197–248, and 262–327);
missense cytoplasmic loop (C-loop) mutations (S2–S3 C-loop, residues 169–196 and S4–S5 C-loop, residues 249–261);
missense N-terminus mutations (residues 1–122);
missense C-terminus mutations (residues 349–676); and
non-missense mutations.
Among the non-missense variants, there were two well-established splicing variants (p.A344A and c.1032+1 G>A) and an in-frame deletion variant (p.339delF) that all affect a portion of the nucleotide sequence of the channel that codes for the KCNQ1 S6 segment. For the sake of clarity, they were also evaluated separately (see Results). Therefore, their association with markers of clinical severity was specifically considered and evaluated in comparison with both the missense p.A341-neighbouring mutations and the other non-missense mutations mapping outside segment S6.
Cellular studies
Cell culture, transfection, and site-directed mutagenesis
See Supplementary material online, Methods Section.
Imaging of membrane expression
Wild-type (WT) and mutant KCNQ1 were tagged with green fluorescent protein (GFP) at the KCNQ1 C-terminus. Chinese hamster ovary (CHO) cells were grown on 8-well chamber slides and transfected using Lipofectamine™ LTX and PLUS™ reagent (Thermo Fisher Scientific). For transfection, 80 ng of WT KCNQ1-GFP or mutant KCNQ1-GFP cDNA and 160 ng of untagged KCNE1 cDNA were used. Forty-eight hours after transfection, cellular localization of KCNQ1 was determined by spinning-disc microscopy (see Supplementary material online, Methods Section).
Protein extraction and western blotting
See Supplementary material online, Methods Section.
Electrophysiological experiments
p.A341V and p.A341-neighbouring mutations p.S338C, p.F339S, p.L342F, p.P343R, p.A344V, and p.G345R were tested. For patch-clamp recordings in WT or homozygous mutant experiments, KCNQ1 (0.25 µg; WT or mutant) and KCNE1 (0.5 µg) cDNA were co-transfected in CHO cells using FuGENE® 6 (Promega Corporation). In studies examining heterozygous conditions, 0.125 μg WT KCNQ1 and 0.125 μg mutant KCNQ1 were transfected with 0.5 μg KCNE1. In addition, yotiao (1 µg) was co-transfected in cells employed for patch-clamp recordings. At 20–24 h after transfection, CHO cells were harvested from the culture dish by a brief trypsinization, washed twice with culture medium, and placed in a perfusion chamber on an inverted microscope that was continuously perfused with an external Tyrode’s solution containing the following (in mmol/L): 132 NaCl, 4.8 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, and 5 glucose (pH = 7.4 with NaOH). Pipettes were filled with solution containing the following (in mmol/L): 110 K-aspartate, 5 ATP-K2, 1 MgCl2, 1 CaCl2, 11 EGTA, and 10 HEPES (pH = 7.3 with KOH). To mimic PKA-mediated β-adrenergic receptor stimulation, 200 µmol/L cAMP and 0.2 μmol/L okadaic acid were added to the pipette solution when appropriate. Experiments were performed using the whole-cell patch-clamp technique at room temperature on the basis of group comparisons (absence or presence of cAMP) for WT, and heterozygous p.A341V and named p.A341-neighbouring variants. All measurements were completed within 5 min of establishing the whole-cell patch to minimize the influence of I Ks rundown.19 Pipette resistance ranged between 1.5 and 3 MΩ, access resistance was determined to be up to 8 MΩ, and the series resistance was electronically compensated at 60–80%. Currents were sampled at 2 kHz after low-pass filtering at 1 kHz. Current density was calculated by dividing current amplitude by cell capacity. Cells were kept at a holding potential of −80 mV. The I/V protocol consisted of 5-ms depolarizing pulses from −50 to +60 mV in 10-mV steps. Tail currents were recorded on repolarization to −40 mV. Voltage dependence of activation (evaluated from normalized I Ks-tail amplitudes) was fitted with a Boltzmann equation: I/I max = 1/{1+exp[(V − V 1/2)/k]} to determine the membrane potential for half-maximal activation (V 1/2) and the slope factor (k).
Statistical analysis
Continuous data were reported as mean and standard deviation or as median and interquartile range (IQR 25th–75th percentile), as appropriate, and analysed by one-way ANOVA or by Kruskal–Wallis test. Categorical variables were presented as absolute and relative frequencies and compared among groups by χ2 or Fisher’s exact test. No Bonferroni adjustments for multiple tests were applied. Event-free survival was described by Kaplan–Meier cumulative estimates, with comparisons performed by the log-rank test. Time from birth to any first cardiac event (syncope, ACA, SCD, whichever occurred first) through age 40 years was considered. To estimate the independent risk of genetic factors of interest for a first cardiac event, a Cox regression analysis was used, stratified by sex to relax the assumption of proportional hazards, and adjusted for QTc duration. As correlation among data points can underestimate the standard error used in significance testing, adjustment was made by using the robust sandwich variance estimator for family relatedness. Hazard ratios (HRs) were presented along with 95% confidence intervals (CIs).
Calculations were performed using the statistical software SPSS Statistics, version 23 (IBM Co, Armonk, NY, USA) and R, version 3.6.28 For statistical comparisons at the cellular level, unpaired two-sided Student’s t-test or one-way ANOVA were used as appropriate. P-value <0.05 was considered statistically significant.
Results
All KCNQ1 mutations identified in the study population are listed in the Supplementary material online, Appendix Table S1, with their respective numbers of carriers and classification. Figure 1 shows their topology along the KCNQ1 channel protein. Of a total of 166 distinct nucleotide sequence variants, most (n = 148, 89%) were classified according to ACMG as pathogenic (n = 79) or likely pathogenic (n = 69), with a minority (n = 18, 11%) as variants of uncertain significance. Furthermore, 113 (68%) are expected to result in missense and 53 (32%) in non-missense amino acid changes. Missense mutations were further subdivided according to location: N-terminus (n = 6; 5%), transmembrane and extracellular loops (n = 44; 39%), C-loops (n = 20; 18%), S6 segment (p.A341V and p.A341-neighbouring mutations; n = 12; 11%), and C-terminus (n = 31; 27%).
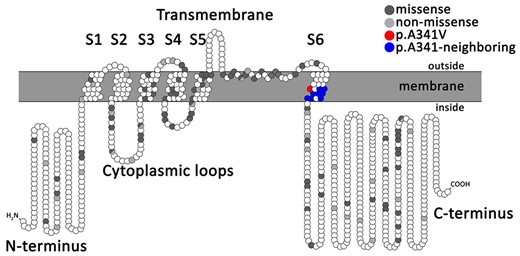
Topology of KCNQ1 missense and non-missense mutations identified in the total study population of 1316 long QT syndrome type 1 patients, according to their amino acid position along the KCNQ1 channel.
Mutations mapping to the S6 segment
Missense p.A341-neighbouring mutations
Table 1 shows the spectrum of all 10 mutations in the KCNQ1 S6 segment around the A341 residue identified in 23 families, with their clinical correlates. Table 2 summarizes the clinical characteristics of all 139 carriers of p.A341-neighbouring mutations, also categorized according to the mutation type (91 missense, 48 non-missense). In full agreement with our previous reports,16 , 18 carriers of the p.A341V mutation (n = 277; 48 families) exhibited the most severe arrhythmia phenotype, regardless of their ethnic background. Their basal QTc was 490 ± 44 ms and the vast majority (73%) was symptomatic for arrhythmic episodes that were life-threatening in 28%. Among the 91 carriers of missense p.A341-neighbouring mutations, basal QTc (477 ± 33 ms) was shorter than that of the p.A341V group (490 ± 44 ms; P = 0.012) and longer than that of the QTc measured in carriers of the other missense LQT1 mutations (467 ± 41 ms; P = 0.037). As shown in Figure 2, there was a similarly lower proportion of subjects with a normal QTc (≤440 ms) among p.A341V and p.A341-neighbouring mutations carriers compared to the rest of the LQT1 group (8% and 12%, respectively, vs. 26%, P < 0.01 for both comparisons); by contrast, as expected from our previous observations,16 the frequency of patients with a QTc exceeding 500 ms remained significantly higher among p.A341V-mutation carriers compared to either of the remaining groups (33% vs. 19% vs. 14%, respectively, P ≤ 0.05 for both comparisons). During a median observation time of 27 (IQR 15–48) years from birth to last contact, 39 (43%) p.A341-neighbouring missense mutation carriers became symptomatic before age 41. This proportion is intermediate among that of the two groups, i.e. 73% and 20%, for p.A341V and the remaining LQT1 carriers of missense mutations, with a similar follow-up time in the three subsets (Table 2). Likewise, when only major arrhythmic events were considered, this frequency in the p.A341-neighbouring mutations carriers (7.7%) was significantly lower than that in the p.A341V subset (28%), but almost two-fold higher than in the other LQT1 patients (4.6%). As expected for all LQT1 symptomatic patients,12 most (91%) of the arrhythmic events among the p.A341-neighbouring mutations carriers occurred during exercise (66%, of which 38% while swimming) or were associated with emotional arousal (25%). Only a few cardiac events occurred at rest or in association with other conditions (9%). Kaplan–Meier curves describing the cumulative survival to any first cardiac event for the p.A341-neighbouring mutations, in comparison with p.A341V and all other pooled LQT1 mutation carriers, are shown in Figure 3. A significant difference in survival was observed (P < 0.001), with the p.A341-neighbouring mutations group confirming its intermediate position among the other two subgroups. The 40-year cumulative event-free survival of p.A341-neighbouring mutation carriers was 49% (95% CI 38–63) compared to 22% (95% CI 17–28) and 76% (95% CI 72–80), respectively. Indeed, they had a significantly (P < 0.001) more favourable prognosis compared to the p.A341V population but remained at a significantly (P < 0.001) higher risk compared to the rest of LQT1 patients. Consistently, at a multivariable Cox model, both S6-segment mutation groups were at a significantly higher risk of a first arrhythmic event compared to all the other missense LQT1 variants (p.A341 neighbouring: HR 2.3, 95% CI 1.6–3.2; P < 0.001; A341V: HR 5.3, 95% CI 3.9−7.2; P < 0.001). A secondary univariate survival analysis performed only on probands to correct for the potential effect of familial clustering on phenotype confirmed the results (Figure 4).
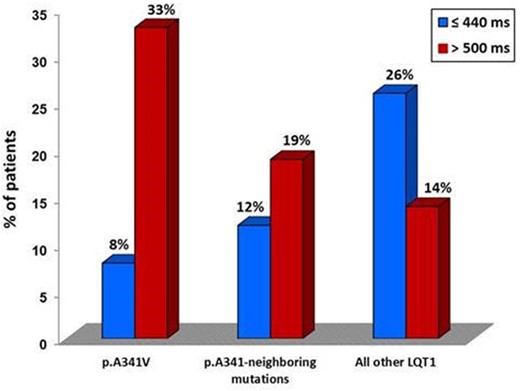
Proportion of missense mutation carriers with a QTc ≤440 and >500 ms in the three study groups. LQT1, long QT syndrome type 1.
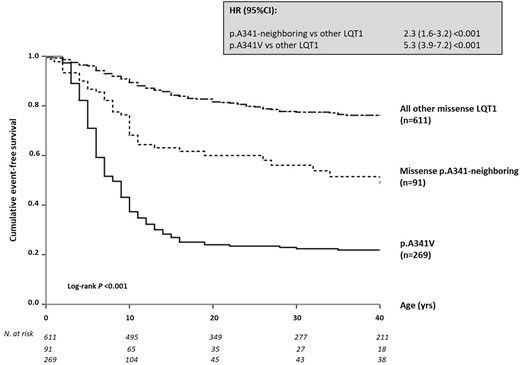
Cumulative event-free survival of patients for all long QT syndrome type 1 missense mutations, i.e. p.A341V, p.A341-neighbouring mutations, and all the other KCNQ1 mutations located outside the S6 region. Top: hazard ratio with 95% confidence interval for the first occurrence of cardiac events in p.A341V and p.A341-neighbouring mutation vs. other missense long QT syndrome type 1 mutation carriers. Bottom: patients at risk per decade of age since birth up to 40 years, according to the genetic subgroup. Discrepancies in numbers of subjects between this figure and Table 2 are due to the lack of precise time at the first event for a few patients. CI, confidence interval; HR, hazard ratio; LQT1, long QT syndrome type 1.
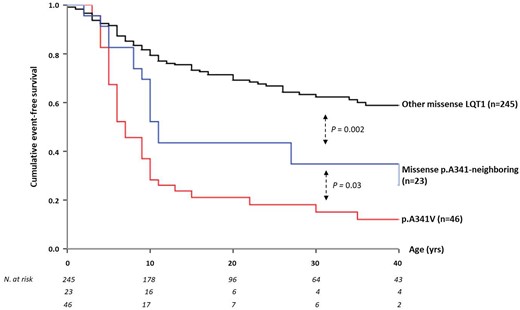
Cumulative event-free survival of genotype-positive probands carrying long QT syndrome type 1 missense mutations: namely, p.A341V, p.A341-neighbouring mutations, and all other KCNQ1 mutations located outside the S6 region. LQT1, long QT syndrome type 1.
Spectrum of p.A341-neighbouring mutations included in the study with the corresponding clinical phenotype
| Genotype-positive subjects | Families | Symptomatic | Possibly asymptomatic | Truly asymptomatic | Major arrhythmic events | |
|---|---|---|---|---|---|---|
| Missense | ||||||
| p.S338C | 2 | 1 | 1 | 1 | 0 | 0 |
| p.F339S | 6 | 2 | 3 | 1 | 2 | 0 |
| p.A341E | 5 | 1 | 4 | 0 | 1 | 2 |
| p.L342F | 14 | 3 | 7 | 3 | 4 | 2 |
| p.P343R | 5 | 2 | 3 | 0 | 2 | 0 |
| p.A344E | 5 | 1 | 2 | 2 | 1 | 0 |
| p.A344V | 31 | 9 | 8 | 8 | 15 | 0 |
| p.G345R | 19 | 2 | 10 | 6 | 3 | 3 |
| p.L347F | 3 | 1 | 0 | 2 | 1 | 0 |
| p.G348D | 1 | 1 | 1 | 0 | 0 | 0 |
| Non-missense | ||||||
| p.339delF | 3 | 2 | 0 | 2 | 1 | 0 |
| p.A344A | 40 | 17 | 23 | 11 | 6 | 7 |
| c.1032 + 1 G>A | 5 | 1 | 4 | 1 | 0 | 0 |
| Total, n (%) | 139 (100) | 43 | 66 (47) | 37 (27) | 36 (26) | 14 (10) |
| Genotype-positive subjects | Families | Symptomatic | Possibly asymptomatic | Truly asymptomatic | Major arrhythmic events | |
|---|---|---|---|---|---|---|
| Missense | ||||||
| p.S338C | 2 | 1 | 1 | 1 | 0 | 0 |
| p.F339S | 6 | 2 | 3 | 1 | 2 | 0 |
| p.A341E | 5 | 1 | 4 | 0 | 1 | 2 |
| p.L342F | 14 | 3 | 7 | 3 | 4 | 2 |
| p.P343R | 5 | 2 | 3 | 0 | 2 | 0 |
| p.A344E | 5 | 1 | 2 | 2 | 1 | 0 |
| p.A344V | 31 | 9 | 8 | 8 | 15 | 0 |
| p.G345R | 19 | 2 | 10 | 6 | 3 | 3 |
| p.L347F | 3 | 1 | 0 | 2 | 1 | 0 |
| p.G348D | 1 | 1 | 1 | 0 | 0 | 0 |
| Non-missense | ||||||
| p.339delF | 3 | 2 | 0 | 2 | 1 | 0 |
| p.A344A | 40 | 17 | 23 | 11 | 6 | 7 |
| c.1032 + 1 G>A | 5 | 1 | 4 | 1 | 0 | 0 |
| Total, n (%) | 139 (100) | 43 | 66 (47) | 37 (27) | 36 (26) | 14 (10) |
Spectrum of p.A341-neighbouring mutations included in the study with the corresponding clinical phenotype
| Genotype-positive subjects | Families | Symptomatic | Possibly asymptomatic | Truly asymptomatic | Major arrhythmic events | |
|---|---|---|---|---|---|---|
| Missense | ||||||
| p.S338C | 2 | 1 | 1 | 1 | 0 | 0 |
| p.F339S | 6 | 2 | 3 | 1 | 2 | 0 |
| p.A341E | 5 | 1 | 4 | 0 | 1 | 2 |
| p.L342F | 14 | 3 | 7 | 3 | 4 | 2 |
| p.P343R | 5 | 2 | 3 | 0 | 2 | 0 |
| p.A344E | 5 | 1 | 2 | 2 | 1 | 0 |
| p.A344V | 31 | 9 | 8 | 8 | 15 | 0 |
| p.G345R | 19 | 2 | 10 | 6 | 3 | 3 |
| p.L347F | 3 | 1 | 0 | 2 | 1 | 0 |
| p.G348D | 1 | 1 | 1 | 0 | 0 | 0 |
| Non-missense | ||||||
| p.339delF | 3 | 2 | 0 | 2 | 1 | 0 |
| p.A344A | 40 | 17 | 23 | 11 | 6 | 7 |
| c.1032 + 1 G>A | 5 | 1 | 4 | 1 | 0 | 0 |
| Total, n (%) | 139 (100) | 43 | 66 (47) | 37 (27) | 36 (26) | 14 (10) |
| Genotype-positive subjects | Families | Symptomatic | Possibly asymptomatic | Truly asymptomatic | Major arrhythmic events | |
|---|---|---|---|---|---|---|
| Missense | ||||||
| p.S338C | 2 | 1 | 1 | 1 | 0 | 0 |
| p.F339S | 6 | 2 | 3 | 1 | 2 | 0 |
| p.A341E | 5 | 1 | 4 | 0 | 1 | 2 |
| p.L342F | 14 | 3 | 7 | 3 | 4 | 2 |
| p.P343R | 5 | 2 | 3 | 0 | 2 | 0 |
| p.A344E | 5 | 1 | 2 | 2 | 1 | 0 |
| p.A344V | 31 | 9 | 8 | 8 | 15 | 0 |
| p.G345R | 19 | 2 | 10 | 6 | 3 | 3 |
| p.L347F | 3 | 1 | 0 | 2 | 1 | 0 |
| p.G348D | 1 | 1 | 1 | 0 | 0 | 0 |
| Non-missense | ||||||
| p.339delF | 3 | 2 | 0 | 2 | 1 | 0 |
| p.A344A | 40 | 17 | 23 | 11 | 6 | 7 |
| c.1032 + 1 G>A | 5 | 1 | 4 | 1 | 0 | 0 |
| Total, n (%) | 139 (100) | 43 | 66 (47) | 37 (27) | 36 (26) | 14 (10) |
Clinical characteristics of all carriers of KCNQ1 mutations according to location (S6 vs. all other gene regions) and coding type (missense vs. non-missense)
| p.A341V (n = 277) | p.A341-neighbouring (n = 139) | All other LQT1 (n = 900) | P-value | |||
|---|---|---|---|---|---|---|
| Missense | Non-missense | Missense | Non-missense | |||
| Patients, n | 277 | 91 | 48 | 614 | 286 | |
| Families, n | 48 | 23 | 20 | 247 | 112 | |
| Females, n (%) | 148 (53) | 49 (54) | 24 (50) | 342 (56) | 169 (59) | 0.615 |
| Follow-up (years),a median (IQR) | 29 (14–52) | 27 (15–48) | 23 (13–45) | 31 (16–48) | 37 (20–49) | 0.08 |
| QTc (ms), mean ± SD (n) | 490 ± 44 (232) | 477 ± 33 (85) | 478 ± 31 (43) | 467 ± 41 (585) | 458 ± 30 (273) | <0.001 |
| ≤440 ms, n (%) | 19 (8) | 10 (12) | 2 (5) | 151 (26) | 82 (30) | <0.001 |
| >500 ms, n (%) | 77 (33) | 16 (19) | 8 (19) | 84 (14) | 15 (5.5) | <0.001 |
| Clinical status, n (%) | <0.001 | |||||
| Symptomatic | 202 (73) | 39 (43) | 27 (56) | 122 (20) | 36 (13) | <0.001 |
| Possibly asymptomatic | 32 (12) | 23 (25) | 14 (29) | 185 (30) | 73 (25) | <0.001 |
| Truly asymptomatic | 43 (15) | 29 (32) | 7 (15) | 307 (50) | 177 (62) | <0.001 |
| Age at first event (years), median (IQR) | 6 (5–9) | 8 (5–11) | 7 (5–11) | 10 (6–17) | 22 (13–28) | <0.001 |
| CA/SCD, n (%) | 78 (28) | 7 (8) | 7 (15) | 28 (5) | 9 (3) | <0.001 |
| p.A341V (n = 277) | p.A341-neighbouring (n = 139) | All other LQT1 (n = 900) | P-value | |||
|---|---|---|---|---|---|---|
| Missense | Non-missense | Missense | Non-missense | |||
| Patients, n | 277 | 91 | 48 | 614 | 286 | |
| Families, n | 48 | 23 | 20 | 247 | 112 | |
| Females, n (%) | 148 (53) | 49 (54) | 24 (50) | 342 (56) | 169 (59) | 0.615 |
| Follow-up (years),a median (IQR) | 29 (14–52) | 27 (15–48) | 23 (13–45) | 31 (16–48) | 37 (20–49) | 0.08 |
| QTc (ms), mean ± SD (n) | 490 ± 44 (232) | 477 ± 33 (85) | 478 ± 31 (43) | 467 ± 41 (585) | 458 ± 30 (273) | <0.001 |
| ≤440 ms, n (%) | 19 (8) | 10 (12) | 2 (5) | 151 (26) | 82 (30) | <0.001 |
| >500 ms, n (%) | 77 (33) | 16 (19) | 8 (19) | 84 (14) | 15 (5.5) | <0.001 |
| Clinical status, n (%) | <0.001 | |||||
| Symptomatic | 202 (73) | 39 (43) | 27 (56) | 122 (20) | 36 (13) | <0.001 |
| Possibly asymptomatic | 32 (12) | 23 (25) | 14 (29) | 185 (30) | 73 (25) | <0.001 |
| Truly asymptomatic | 43 (15) | 29 (32) | 7 (15) | 307 (50) | 177 (62) | <0.001 |
| Age at first event (years), median (IQR) | 6 (5–9) | 8 (5–11) | 7 (5–11) | 10 (6–17) | 22 (13–28) | <0.001 |
| CA/SCD, n (%) | 78 (28) | 7 (8) | 7 (15) | 28 (5) | 9 (3) | <0.001 |
CA, cardiac arrest; IQR, interquartile range; LQT1, long QT syndrome type 1; SCD, sudden cardiac death; SD, standard deviation.
From birth to last contact.
Clinical characteristics of all carriers of KCNQ1 mutations according to location (S6 vs. all other gene regions) and coding type (missense vs. non-missense)
| p.A341V (n = 277) | p.A341-neighbouring (n = 139) | All other LQT1 (n = 900) | P-value | |||
|---|---|---|---|---|---|---|
| Missense | Non-missense | Missense | Non-missense | |||
| Patients, n | 277 | 91 | 48 | 614 | 286 | |
| Families, n | 48 | 23 | 20 | 247 | 112 | |
| Females, n (%) | 148 (53) | 49 (54) | 24 (50) | 342 (56) | 169 (59) | 0.615 |
| Follow-up (years),a median (IQR) | 29 (14–52) | 27 (15–48) | 23 (13–45) | 31 (16–48) | 37 (20–49) | 0.08 |
| QTc (ms), mean ± SD (n) | 490 ± 44 (232) | 477 ± 33 (85) | 478 ± 31 (43) | 467 ± 41 (585) | 458 ± 30 (273) | <0.001 |
| ≤440 ms, n (%) | 19 (8) | 10 (12) | 2 (5) | 151 (26) | 82 (30) | <0.001 |
| >500 ms, n (%) | 77 (33) | 16 (19) | 8 (19) | 84 (14) | 15 (5.5) | <0.001 |
| Clinical status, n (%) | <0.001 | |||||
| Symptomatic | 202 (73) | 39 (43) | 27 (56) | 122 (20) | 36 (13) | <0.001 |
| Possibly asymptomatic | 32 (12) | 23 (25) | 14 (29) | 185 (30) | 73 (25) | <0.001 |
| Truly asymptomatic | 43 (15) | 29 (32) | 7 (15) | 307 (50) | 177 (62) | <0.001 |
| Age at first event (years), median (IQR) | 6 (5–9) | 8 (5–11) | 7 (5–11) | 10 (6–17) | 22 (13–28) | <0.001 |
| CA/SCD, n (%) | 78 (28) | 7 (8) | 7 (15) | 28 (5) | 9 (3) | <0.001 |
| p.A341V (n = 277) | p.A341-neighbouring (n = 139) | All other LQT1 (n = 900) | P-value | |||
|---|---|---|---|---|---|---|
| Missense | Non-missense | Missense | Non-missense | |||
| Patients, n | 277 | 91 | 48 | 614 | 286 | |
| Families, n | 48 | 23 | 20 | 247 | 112 | |
| Females, n (%) | 148 (53) | 49 (54) | 24 (50) | 342 (56) | 169 (59) | 0.615 |
| Follow-up (years),a median (IQR) | 29 (14–52) | 27 (15–48) | 23 (13–45) | 31 (16–48) | 37 (20–49) | 0.08 |
| QTc (ms), mean ± SD (n) | 490 ± 44 (232) | 477 ± 33 (85) | 478 ± 31 (43) | 467 ± 41 (585) | 458 ± 30 (273) | <0.001 |
| ≤440 ms, n (%) | 19 (8) | 10 (12) | 2 (5) | 151 (26) | 82 (30) | <0.001 |
| >500 ms, n (%) | 77 (33) | 16 (19) | 8 (19) | 84 (14) | 15 (5.5) | <0.001 |
| Clinical status, n (%) | <0.001 | |||||
| Symptomatic | 202 (73) | 39 (43) | 27 (56) | 122 (20) | 36 (13) | <0.001 |
| Possibly asymptomatic | 32 (12) | 23 (25) | 14 (29) | 185 (30) | 73 (25) | <0.001 |
| Truly asymptomatic | 43 (15) | 29 (32) | 7 (15) | 307 (50) | 177 (62) | <0.001 |
| Age at first event (years), median (IQR) | 6 (5–9) | 8 (5–11) | 7 (5–11) | 10 (6–17) | 22 (13–28) | <0.001 |
| CA/SCD, n (%) | 78 (28) | 7 (8) | 7 (15) | 28 (5) | 9 (3) | <0.001 |
CA, cardiac arrest; IQR, interquartile range; LQT1, long QT syndrome type 1; SCD, sudden cardiac death; SD, standard deviation.
From birth to last contact.
Non-missense mutations mapping to S6
In 48 patients from 20 families, there were three non-missense mutations (the p.339delF in-frame deletion, and the well-known p.A344A and c.1032+1 G>A splicing variants) that affect the nucleotide sequence of the channel at a region that codes for part of the KCNQ1 S6 segment (Table 1). Although in general non-missense mutations are largely expected to produce aberrant transcripts that will undergo early degradation and will not reach the cell membrane to confer dominant-negative effects on WT channels,14 available functional studies for p.339delF29 and p.A344A30 have previously shown that these actually act through dominant-negative mechanisms and for this reason they were also analysed separately (Table 2 and Figure 4). As within this group the hot spot A344A pathogenetic splicing variant predominates with 40 of 48 cases (83%), we looked at it in isolation. We found that 23 (57.5%) patients with this common mutation are symptomatic, an observation in agreement with what reported by a Japanese study.31 Interestingly, altogether the three non-missense mutations behaved similarly to the 10 missense (p.A341-neighbouring) mutations for both QTc (478 ± 31 vs. 477 ± 33 ms) and cardiac event rates, with a 40-year cumulative event-free survival of 32% (95% CI 19−55) vs. 49% (95% CI 38−63) (log-rank P = 0.054). By contrast, compared to carriers of all other non-missense variants located anywhere else, they carried a much higher risk of arrhythmic events, with a 40-year cumulative survival of 32% (95% CI 19−55) vs. 83% (95% CI 78−88) (P < 0.001; Figure 5).
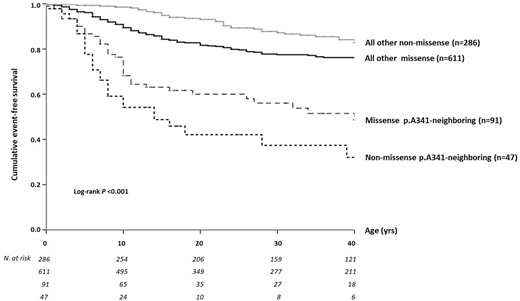
Cumulative event-free survival of patients with p.A341V-neighbouring mutations—either missense or non-missense—compared with all other variants elsewhere located in KCNQ1, also by mutation type.
Mutations outside the S6 segment
Clinical outcome of S6 mutations vs. all other KCNQ1 mutations
Tables 2 and 3 summarize the clinical characteristics of the 900 carriers of all other (non-S6 segment) mutations. Missense mutations involving membrane-spanning segments, including the pore region of KCNQ1, carried the highest risk for arrhythmic events (31%), compared to N- and C-termini and non-missense mutations (5%, 13%, and 13%, respectively). Interestingly, among the 20 different mutations identified in the C-loops S2–S3 and S4–S5, the 38 carriers of three variants, i.e. p.G189R, p.R190Q, and p.V254M, which were previously associated with a defective PKA-dependent I Ks stimulation,15 showed a significantly longer QTc (491 ± 46 ms) compared to that of the remaining 17 C-loop mutation carriers (470 ± 43 ms), and also of all other KCNQ1 genetic subgroups (Table 3; P < 0.01 for all comparisons). However, the difference in the proportion of symptomatic patients between the two C-loop subgroups, classified according to a known defective PKA-dependent I Ks regulation, was moderate (21% vs. 15%) and non-significant.
Clinical status and basal QTc in all other KCNQ1 mutation carriers according to mutation topology
| Mutation | No. | Symptomatic | Possibly asymptomatic | Truly asymptomatic | QTca |
|---|---|---|---|---|---|
| Non-missense | 286 | 36 (13%) | 73 (25%) | 177 (62%) | 458 ± 30 |
| C-loop | 184 | 27 (15%) | 44 (24%) | 113 (61%) | 470 ± 43 |
| C-loop with defective I Ks regulation | 38 | 8 (21%) | 11 (29%) | 19 (50%) | 491 ± 46 |
| Transmembrane | 211 | 66 (31%) | 57 (27%) | 88 (42%) | 467 ± 40 |
| C-terminus | 142 | 19 (13%) | 59 (42%) | 64 (45%) | 463 ± 36 |
| N-terminus | 39 | 2 (5%) | 14 (36%) | 23 (59%) | 452 ± 37 |
| Total | 900 | 158 (17%) | 258 (29%) | 484 (54%) |
| Mutation | No. | Symptomatic | Possibly asymptomatic | Truly asymptomatic | QTca |
|---|---|---|---|---|---|
| Non-missense | 286 | 36 (13%) | 73 (25%) | 177 (62%) | 458 ± 30 |
| C-loop | 184 | 27 (15%) | 44 (24%) | 113 (61%) | 470 ± 43 |
| C-loop with defective I Ks regulation | 38 | 8 (21%) | 11 (29%) | 19 (50%) | 491 ± 46 |
| Transmembrane | 211 | 66 (31%) | 57 (27%) | 88 (42%) | 467 ± 40 |
| C-terminus | 142 | 19 (13%) | 59 (42%) | 64 (45%) | 463 ± 36 |
| N-terminus | 39 | 2 (5%) | 14 (36%) | 23 (59%) | 452 ± 37 |
| Total | 900 | 158 (17%) | 258 (29%) | 484 (54%) |
QTc measurements are mean ± standard deviation.
Clinical status and basal QTc in all other KCNQ1 mutation carriers according to mutation topology
| Mutation | No. | Symptomatic | Possibly asymptomatic | Truly asymptomatic | QTca |
|---|---|---|---|---|---|
| Non-missense | 286 | 36 (13%) | 73 (25%) | 177 (62%) | 458 ± 30 |
| C-loop | 184 | 27 (15%) | 44 (24%) | 113 (61%) | 470 ± 43 |
| C-loop with defective I Ks regulation | 38 | 8 (21%) | 11 (29%) | 19 (50%) | 491 ± 46 |
| Transmembrane | 211 | 66 (31%) | 57 (27%) | 88 (42%) | 467 ± 40 |
| C-terminus | 142 | 19 (13%) | 59 (42%) | 64 (45%) | 463 ± 36 |
| N-terminus | 39 | 2 (5%) | 14 (36%) | 23 (59%) | 452 ± 37 |
| Total | 900 | 158 (17%) | 258 (29%) | 484 (54%) |
| Mutation | No. | Symptomatic | Possibly asymptomatic | Truly asymptomatic | QTca |
|---|---|---|---|---|---|
| Non-missense | 286 | 36 (13%) | 73 (25%) | 177 (62%) | 458 ± 30 |
| C-loop | 184 | 27 (15%) | 44 (24%) | 113 (61%) | 470 ± 43 |
| C-loop with defective I Ks regulation | 38 | 8 (21%) | 11 (29%) | 19 (50%) | 491 ± 46 |
| Transmembrane | 211 | 66 (31%) | 57 (27%) | 88 (42%) | 467 ± 40 |
| C-terminus | 142 | 19 (13%) | 59 (42%) | 64 (45%) | 463 ± 36 |
| N-terminus | 39 | 2 (5%) | 14 (36%) | 23 (59%) | 452 ± 37 |
| Total | 900 | 158 (17%) | 258 (29%) | 484 (54%) |
QTc measurements are mean ± standard deviation.
The comparative 40-year cumulative survival to any first cardiac event is shown in Figure 6 for all the predefined subsets of KCNQ1 mutation carriers according to type and location of the identified genetic variants. All the p.A341-neighbouring mutations (missense and non-missense analysed separately) confirmed their intermediate position among the most severe p.A341V group and the relatively more benign groups represented by non-missense, C/N-terminus, C-loop mutations, and also all other membrane-spanning mutations. Findings from a Cox regression analysis assessing the effect of distinct groups of mutations on outcome are summarized in Supplementary material online, Table S2.
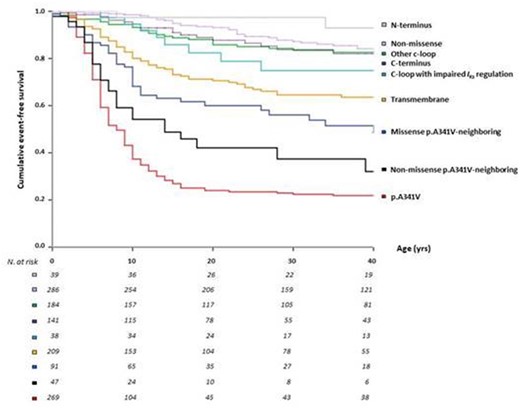
Cumulative event-free survival of patients for all mutations per KCNQ1 region, i.e. p.A341V, p.A341V-neighbouring mutations, other membrane-spanning segments (transmembrane), C-loops (S2–S3 and S4–S5), also by protein kinase A-dependent I Ks-channel activation, C/N termini, and non-missense variants.
β-Blocker therapy
Adequate information on therapy (either β-blocker or no therapy) was available for 960 (73%) LQT1 subjects, including 233/277 (84%) p.A341V carriers, 80/91(88%) missense p.A341 neighbouring mutation carriers, and 411/614 (67%) genotype-positive individuals for other missense mutations elsewhere located along KCNQ1 (Supplementary material online, Appendix Table S3). Of the 80 missense p.A341 neighbouring mutation carriers analysed, 50 (62.5%) were on β-blocker therapy and, during a follow-up time of 11 years (IQR 4−24), 7 (14%) patients had at least one cardiac event, which in four cases was lethal (SCD n = 3) or near-lethal (ACA n = 1). The incidence of breakthrough events despite β-blockers among these p.A341-neighbouring mutations carriers was significantly lower compared to that among p.A341V carriers (14% vs. 52%, P < 0.001) and higher compared to the remaining LQT1 patients (14% vs. 5%, P = 0.03). Syncope on β-blockers, but not ACA, SCD, and ICD shocks, was more frequent among carriers of non-missense than missense p.A341 neighbouring mutations (30% vs. 14%).
Cellular studies
In line with previous work by us19 and others,15 we investigated the cellular underpinning of our clinical findings by assessing the PKA-dependent regulation of mutant I Ks for the KCNQ1 S6 region of which carriers exhibited the most severe clinical phenotype, p.A341V vs. p.A341-neighbouring mutations. First, we determined the effect of mutations on homozygous KCNQ1-membrane expression by confocal microscopy in CHO cells co-transfected with GFP-tagged KCNQ1 mutant or WT and KCNE1 subunits. Our results show (Supplementary material online, Appendix Figure S1) that p.A341V19 and p.A341-neighbouring mutations p.S338C, p.A344V, and p.G345R had a clear fluorescent membrane expression, but not p.F339S, p.L342F, and p.P343R. These results were grossly corroborated by Western blot experiments in which the cytoplasmic (whole-lysate) and membrane-fraction expressions of these KCNQ1 mutations were compared with WT channels (Supplementary material online, Appendix Figure S1B and C). Homozygous expression led to a measurable (although significantly reduced) I Ks for p.A341V and all p.A341-neighbouring mutations (Supplementary material online, Appendix Figure S1D).
Mutant I Ks was subsequently characterized in the absence or presence of cAMP stimulation (Figure 7). To this end, we mimicked the heterozygous status of carriers by co-transfecting WT KCNQ1, mutant KCNQ1 and KCNE1 subunits in a 1:1:3 molar ratio. Significant suppression of unstimulated I Ks was found for p.A341V and p.A341-neighbouring mutations p.L342F, p.P343R, and p.G345R, at a variable degree. Kinetic differences are indicated in Supplementary material online, Appendix Table S3. To mimic β-adrenergic receptor stimulation in WT and mutant I Ks, cAMP/okadaic acid was added to the pipette solution in a group comparison vs. unstimulated current. Unlike what was reported for p.A341V,19 I Ks tails increased significantly upon cAMP/okadaic acid for all p.A341-neighbouring mutations (Figure 7B and D), with a leftward shift of their activation curves in most cases, indicating significant responsiveness to the β-adrenergic receptor stimulation (for kinetic parameters see Supplementary material online, Appendix Table S3). Thus, among the p.A341-neighbouring mutations mapping to the S6 segment, impairment of cAMP-dependent I Ks stimulation was demonstrated only for p.A341V, the most malignant mutation.

Protein kinase A-dependent regulation of mutant I Ks for p.A341V vs. p.A341-neigboring mutations. (A) Localization of p.A341V (red) and A341-neighbouring mutations (blue) on S6 in KCNQ1. (B) Averaged I Ks-density traces for wild type, p.S338C, p.F339S, p.A341V, p.L342F, p.P343R, p.A344V, and p.G345R, all heterozygously expressed in the absence (n = 11, 8, 8, 9, 19, 6, 11, and 21 cells, respectively) or presence (+; n = 15, 6, 9, 12, 16, 8, 6, and 14 cells, respectively) of intrapipette cyclic adenosine monophosphate/okadaic acid during a 5-s depolarizing pulse to +60 mV (holding potential −80 mV). Blue arrows indicate increased tail current density; red arrow indicates lack of tail current enhancement. (C) I Ks-tail densities for wild type and all heterozygously expressed mutations in the absence of cyclic adenosine monophosphate. Multigroup comparison using one-way ANOVA for wild type vs. p.S338C, P = 0.325; vs. p.F339S, P = 0.637; vs. p.A341V, P = 0.018; vs. p.L342F, P = 0.008; vs. p.P343R, P = 0.0001; vs. p.A344V, P = 0.420; and vs. p.G345R, P = 0.001. (D) Relative tail current densities compared to normalized values without cyclic adenosine monophosphate. All mutant channels responded to cyclic adenosine monophosphate/okadaic acid, except for p.A341Vhet. Statistical comparison between the absence and presence of cyclic adenosine monophosphate/okadaic acid: wild type, P = 0.034; p.S338C, P = 0.018; p.F339S, P = 0.031; p.A341V, P = 0.202; p.L342F, P = 0.015; p.P343R, P = 0.020; p.A344V, P = 0.036; and p.G345R, P = 0.022. *Statistically significant difference. cAMP/OA, cyclic adenosine monophosphate/okadaic acid; WT, wild type.
Discussion
The unprecedented size of our LQT1 population (>1300 patients) provides a solid basis for our findings. We demonstrate that KCNQ1 p.A341-neighbouring mutations in the S6 segment of the channel are associated with a more deleterious arrhythmia phenotype and a poorer cumulative survival than mutations from any other region. While the p.A341V mutation stands out in severity, as we have reported,16–18 largely due to its loss of PKA-dependent I Ks regulation, all other p.A341-neighbouring mutations tested show an almost normal I Ks enhancement. Nonetheless, they do correlate with a more severe phenotype compared to all other LQT1 mutations (although not due to PKA insensitivity), thus providing definitive evidence for the importance of mutation location in determining the specific genotype–phenotype correlation. This further refines risk stratification for LQTS patients, representing another step towards precision medicine.5 , 32
The present vs. previous studies
Studies aiming to correlate genotype and phenotype were already attempted soon after the identification of the main LQTS genes in 1995–96. In 1997, Donger et al.33 were the first to introduce the notion of ‘forme fruste’ LQTS due to a C-terminal KCNQ1 mutation, which was associated with a much milder phenotype (i.e. less prolonged QTc, less symptoms, and less SCD events) in carrier patients with respect to patients carrying mutations in the pore and S6 segment, as well as the S4–S5 C-loop. The seed for the concepts of ‘mutation topology’ or ‘location’ was thus planted. The game-changer came 10 years later when Moss et al.14 subgrouped the 77 unique KCNQ1 mutations identified in 600 LQT1 patients based on location (transmembrane vs. N/C terminal), coding type (missense vs. non-missense), and biophysical effect (dominant-negative vs. haploinsufficient). They demonstrated that patients with transmembrane, missense, or dominant-negative mutations were at significantly increased risk compared to patients carrying C-terminal, non-missense, or haploinsufficient mutations, independently of traditional clinical risk factors and β-blocker therapy.14
These results made sense from a biological standpoint. The mechanistic basis by which KCNQ1 mutations confer their effects and result in I Ks reduction was initially broadly categorized under the umbrella terms ‘haploinsufficiency’ and ‘dominant-negative suppression’.14 Since the functional I Ks channel is a tetrameric protein, these two mechanisms could explain the potential contribution, or not, of mutant subunits to the tetrameric channel complex, as well as the respective functional consequences. Haploinsufficient KCNQ1 mutations are those that do not interfere with the function of the WT channel product, nor do their products form heterotetrameric channels and generally result in 50% of WT channels being available at the plasma membrane and conducting I Ks currents. This is a more favourable scenario than dominant-negative mutations, which may exert their effects by either co-assembling and retaining WT subunits intracellularly, thus reducing dramatically I Ks, or form heterotetrameric channels at the cell membrane with much altered biophysical properties.14 , 29 , 30 , 34 This implies that dominant-negative mutations may introduce changes to the channel complex that are structurally less disruptive, thus permitting their co-assembly with WT subunits, while as the number of subunits in a complex increases to form the functional complex, so does the potential of a dominant-negative effect.35 In either case, the overall effect is a >50% I Ks reduction and, depending on each particular mutation, this reduction may range from mild (>50%) to significant (≤93.75%).14 , 34 , 35
While missense mutations may have diverse and unpredictable functional consequences, non-missense mutations are considered more likely to cause haploinsufficiency, since they result in aberrant transcripts that may easily be subjected to early intracellular degradation due to nonsense mediated mRNA decay.14 , 34 However, there are notable exceptions, such as a number of KCNQ1 non-missense mutations resulting in mutant subunits that are nevertheless able to form heterotetramers with their WT counterparts and cause dominant-negative suppression of WT channels. This has been demonstrated by in vitro functional studies for diverse KCNQ1 non-missense mutations, such as in-frame deletions29 and splicing30 and frameshift36 mutations, two of which form part of the non-missense mutations mapping to the S6 segment in this study. In addition, several missense mutations have been shown to act through more than one mechanism.37 , 38 This highlights the complexities of accurately portraying, and even more predicting, the biophysical effects of each mutation in the complex cellular environment.
The KCNQ1-p.A341V pathogenic variant was first functionally characterized in Xenopus oocytes in the late 1990s by Sanguinetti’s group.39 They showed that co-expression of A341V and WT KCNQ1 caused only a small, but statistically significant, rightward shift in the voltage dependence of I Ks activation and they concluded that the mutation conferred ‘little or no dominant-negative effect’.39 , 40 Another study concluded that p.A341V is a simple loss-of-function mutation without dominant-negative effects.41 These in vitro results, surprisingly, did not correlate with the severity of the clinical phenotypes of the respective p.A341V-positive patients, but this was attributed to other unknown factors.39 When we identified the KCNQ1-p.A341V founder mutation in a South African population where we observed a particularly malignant clinical expression of LQTS, we were also puzzled by this lack of genotype–phenotype correlation.16 Consequently, we decided to reassess functionally the p.A341V variant in CHO cells, thus eliminating many of the confounding factors of previously used assays,16 although we cannot completely exclude that some cells selected for electrophysiological experiments did not express one or more of the transfected plasmids.
We found that p.A341V exerts a dominant-negative effect on I Ks, albeit milder than that of a typical dominant-negative mutation, which was more in line with the clinical phenotype.16–18 Still, the high arrhythmic potential of KCNQ1-p.A341V could not be solely explained by being missense, transmembrane, and with a mild dominant-negative effect, especially when compared to other KCNQ1 mutations with similar features.18 In experiments similar to the present ones, we mimicked the conditions of PKA-mediated β-adrenergic receptor stimulation and showed that p.A341V, beyond affecting basal I Ks, markedly cripples I Ks under conditions of β-adrenergic stimulation,19 an experimental observation that was for the first time much more in accordance with the ECG changes and the clinical features observed in p.A341V-positive patients. The observation that this pathogenic variant exerts its effects in a manner much more complex than what could be explained by simply dichotomizing it as missense or non-missense, transmembrane or N/C terminal, haploinsufficient, or dominant-negative, led us to focus our attention on the region of the S6 transmembrane channel segment where the p.A341V resides.
At variance with previous studies that have lumped together all transmembrane mutations, we used p.A341V as a flag, to map a region possibly worth being explored and analysed separately, i.e. the S6-transmembrane segment. By doing so, we have identified the p.A341-neighbouring mutations as conferring a more severe arrhythmic phenotype compared to all other LQT1 mutations, albeit lower than that conferred by p.A341V itself. Barsheshet et al.15 previously reported a higher risk of life-threatening arrhythmic events among carriers of C-loop mutations compared to other predefined KCNQ1 genetic subgroups. Our findings vary with those of that study.15 There are several factors that may account for this difference. First, we were able to analyse both the p.A341V and the p.A341-neighbouring mutations as separate subgroups instead of lumping them together in the transmembrane subgroup.13 Considering the relatively low overall risk profile of their 376 patients with membrane-spanning mutations,15 the severity effect conferred by only 22 (6%) p.A341V patients could have easily been diluted. Conversely, the number of subjects with C-loop mutations is comparable between the two studies (17% vs. 15%15) Also, it is not clear whether the p.R243C mutation (10% of C-loop mutation carriers in Barsheshet’s study15) can be safely classified as a C-loop mutation or S4 transmembrane, since discrepancies exist between topological (UniProtKB—P51787)27 and structural models.42 Although the clinical severity of the 13 p.R243C patients remained unclear from that paper,15 the cellular finding of a loss of PKA-dependent I Ks regulation for p.R243C15 suggests a positive association with arrhythmic events and potentially an overestimation of the severity of the C-loop subgroup. In our study, with a fair representation of mutations mapping to different subdomains along the channel protein, the S6 p.A341V mutation, primarily, and its S6 neighbours, secondarily, emerge as significantly associated with increased arrhythmic risk compared to all other LQT1 mutations, thus highlighting the importance of this segment in channel function.
The importance of position A341 and its neighbours in the S6 segment
The unusual clinical severity of KCNQ1-p.A341V has been studied in detail.16 , 18 In the present study, 73% of the 277 p.A341V carriers manifested syncope, cardiac arrest, or sudden death before the age of 41 years, with first symptoms occurring early in life and with QTc being markedly prolonged. The malignant p.A341V mutation is missense, transmembrane and causes a mild dominant-negative suppression of basal I Ks , 16 , 18 but at the same time, it confers both loss of basal I Ks and loss of PKA-dependent I Ks stimulation.19 We hypothesized that the p.A341V-neighbouring mutations associated with a more severe phenotype, albeit less than p.A341V itself, could also confer some degree of impaired I Ks regulation, but our functional study of six such mutations did not demonstrate a similar underlying mechanism. As it happens in science, despite the strength of the evidence, not always a novel finding is fully explained and a correct explanation may come subsequently, as science progresses. Although our results do not directly demonstrate why p.A341-neighbouring mutations confer a higher arrhythmic risk than most other KCNQ1 mutations, some tenable hypotheses can be made based on previous functional studies of the S6 segment. This segment plays critically important roles in two fundamental processes: KCNQ1 channel gating and modulation. It is well conserved among the families of voltage-gated K+ channels and exhibits a very low rate of evolution, with an enrichment for dysfunctional genetic variants.43 In the tetrameric structure of the channel, voltage-gated activation likely results from the simultaneous bending of all S6 segments away from the central axis, enlarging the opening of the pore to permit the diffusion of K+ ions. Within S6 lie specific amino acid motifs, such as the ‘PAG’ (residues P343, A344, G345) motif, that act as hinges, thus allowing a dynamic S6 movement.44–46 Just a couple of residues downstream, S6 also contains a ‘GSG’ motif (G348, S349, G350) that naturally restricts the pore.42 , 46 Upon S6 bending and expansion, the pore dilates to a wider radius, accommodating ion conduction.
The S6 segment also plays a role in channel modulation through its direct interaction or proximal contact with the channel’s accessory subunits.47 , 48 Mutations p.S338C and p.F340C have been in fact shown to alter KCNQ1 modulation by KCNE1 and KCNE3, respectively,47 while KCNE1 has been shown to naturally interact with the KCNQ1 pore region through residues F339 and F340.48 More recently, transmembrane interactions of KCNE3 and KCNQ1 were specifically mapped within a cleft, partially formed by the contribution of the S6 segment from one subunit.46 Here, under what we have defined as p.A341-neighbouring mutations, 8 of the 10 unique missense mutations in 68 out of 86 carrier patients actually affect the specific residues of the above-mentioned motifs (Supplementary material online, Table S1), thus supporting the importance of this segment for normal channel function, and further justify the focus of our study.
Clinical and conceptual implications
Our data stress the importance of mutation location within the S6 transmembrane segment of the KCNQ1 channel protein. From a clinical point of view, this type of information should not be ignored and should usefully complement a progressively more accurate risk stratification together with the implications of the presence/absence of ‘modifier genes’.6 , 49–53 LQT1 carriers of a mutation located in the S6 segment should be now regarded as at higher risk and be treated more aggressively (at the very least with full-dose β-blockade and frequent 12-lead Holter recordings to monitor repolarization changes). From a conceptual point of view, the present study highlights the growing importance of coupling carefully collected phenotypic information with comprehensive genetic analyses, advanced cellular electrophysiology, and eventually also insight from structure–function relationships of normal channel function, as a critical way forward for the implementation of precision medicine in channelopathies.5 , 32 In addition, physiologically more relevant in vitro systems, such as patient-specific induced pluripotent stem cell-derived cardiomyocytes, could be useful for validating previous findings as well as for providing insight into the expression levels of transcripts resulting from missense vs. non-missense mutations.32 , 54 Similarly, the recent availability of refined three-dimensional structural models of the human KCNQ1 channel protein may advance the understanding of how mutations impair normal channel structure-function relationships.42 , 46
Clinical cardiologists should become aware that a proper clinical management of their LQTS patients can no longer ignore, as a minimum, a reasonable level of understanding of the implications of the main characteristics of the disease-causing mutations, including their type and location.
Supplementary material
Supplementary material is available at European Heart Journal online.
Acknowledgements
The authors thank Kèvin Knoops, PhD, Maastricht MultiModal Molecular Imaging Institute (M4I), Maastricht University, The Netherlands, for valuable advice on the spinning-disc microscopy experiments; Ida Skrinde Leren, MD, PhD, Christine Rootwelt, MD, Rukijat A. Ildarova, MD, and Althea Goosen, RN, for contributing to the data collection and follow-up; Luca Sala, PhD, for helpful discussions on aspects of cellular electrophysiology; and Valerie Corfield, PhD, for critical contributions to the early studies of p.A341V. The authors are also grateful to Pinuccia De Tomasi, BS, for expert editorial support. The structural image of the KCNQ1 channel in the Graphical abstract was created using the structural model of the human KCNQ1 channel protein (Protein Databank accession no. 6UZZ) with the support of the VMD molecular visualization program. VMD is developed with NIH support by the Theoretical and Computational Biophysics group at the Beckman Institute, University of Illinois at Urbana-Champaign, IL, USA.
Funding
P.G.A.V. was supported by The Netherlands CardioVascular Research Initiative (CVON PREDICT2, grant 2018-30), Den Haag, The Netherlands, and the Health Foundation Limburg, Maastricht, The Netherlands. P.J.S. and L.C. were partially supported by Leducq Foundation for Cardiovascular Research grant 18CVD05 ‘Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies’; by ERA-CVD JTC-2018-026 grant ‘Electromechanical presages of sudden cardiac death in the young: integrating imaging, modelling and genetics for patient stratification’, and by EJP RD Joint Transnational Call 2019 grant “LQTS-NEXT: To the NEXT level of risk prediction in patients with Long QT Syndrome”.
Conflict of interest: none declared.
Data availability
The data underlying this article are available in the article itself and in its online supplementary material, or will be shared upon reasonable request to the corresponding author.
References
R Core Team.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}