





This editorial refers to ‘Acute mental stress drives vascular inflammation and promotes plaque destabilization in mouse atherosclerosis’, by J. Hinterdobler et al., https://doi.org/10.1093/eurheartj/ehab371.
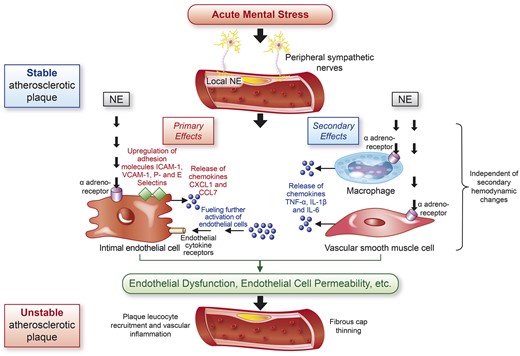
Diagram outlining the proposed pathophysiological mechanisms by which acute mental stress leads to unstable atherosclerotic plaque and cardiovascular events. Acute mental stress leads to the stimulation of peripheral sympathetic nerves, increasing locally available norepinephrine. Norepinephrine exerts primary effects on endothelial cells via α-adrenoreceptors, leading to up-regulation of adhesion molecules and release of chemokines. These effects are secondarily enhanced via the binding of norepinephrine to surface receptors of macrophages and vascular smooth muscle cells that release chemokines that further stimulate the effector endothelial cells. These effects collectively lead to leucocyte recruitment and adhesion. Endotheial dysfunction and increased endothelial cell permeability results in vascular inflammation, fibrous cap thinning, plaque instability, and ensuing cardiovascular events. CXCL1, chemokine C–X–C motif ligand 1; CCL7, chemokine C–C motif ligand 7; NE, norepinephrine; ICAM-1, intracellular adhesion molecule-1; VCAM-1, vascular adhesion molecule-1.
A role for mental stress (MS) as a risk factor for cardiovascular disease (CVD) continues to emerge. Previous studies demonstrated that acute MS is associated with an increased risk of myocardial infarction (MI), sudden cardiac death, and stroke, and may trigger events in individuals with a high pre-existing atherosclerotic plaque burden.1 However, less is known about the underlying mechanisms of this relationship, and therefore what treatments could be harnessed to reduce MS-related risk.
Hinterdobler et al.2 have made a valuable contribution to our understanding of the mechanisms that relate acute MS to CVD events. In their study, published in this issue of the European Heart Journal, the authors demonstrated rapid depletion of peripherally circulating leucocytes from the blood of humans and mice after exposure to acute MS. To uncover the target of these cells, they used novel cell-tracking experiments in murine models exposed to MS in the form of restraint/immobilization, and showed that inflammatory leucocytes, specifically monocytes and neutrophils, were promptly taken up by specific tissues including the heart, lung, and skin. In atherosclerosis prone mice, acute MS led to an expanded inflammatory leucocyte pool in atherosclerotic plaque. This link has previously been suggested in the clinical setting with the help of sophisticated imaging modalities. Using [18F]fluorodeoxyglucose ([18F]FDG) positron emission tomography/computed tomography (PET/CT), amygdala activity, a marker of emotional stress, carotid arterial inflammation, and macrophage haematopoiesis were found to be significantly elevated in patients with a recent MI compared with controls,3 advancing the notion that MS-associated neurobiological effects lead to CVD events through acute plaque destabilization. That study, however, raised important questions about causality and direction of effect. While MS and its related neurobiological activity could lead to vascular dysfunction, inflammation, vulnerable plaque, and MI, it is also plausible that the experience of MI led to greater MS and its associated neurobiological activity. In such an instance, causality could be implied potentially by demonstrating vascular inflammation specifically at the site of infarction. In another study, the cause vs. effect question was partly addressed, whereby [18F]FDG PET/CT-assessed amygdala activity was associated with an increased frequency of CVD events at follow-up, even after adjusting for CVD risk factors and pre-existing atherosclerotic disease.4 The link between amygdala activity and CVD events appeared to be mediated by increased arterial inflammation, which in turn was mediated by up-regulated bone marrow activity. Yet, the successive temporal link between MS and de novo vascular inflammation and plaque rupture remains to be demonstrated.
Hinterdobler et al. also noted that atherosclerotic plaques that showed increased uptake of inflammatory cells in response to MS also had features associated with thinning of the fibrous cap, characterized by more matrix metalloproteinase and fewer procollagen transcripts. High plaque leucocyte content and shrinking fibrous caps increase the risk of plaque rupture,5 and indeed the authors went on to demonstrate histological evidence in a plaque rupture model that occurred more often after acute MS exposure. This important finding connects the upstream MS with the downstream inciting pathological event, and provides greater insight into the directionality question in an animal model. An important next step would be extending these findings to humans, potentially using coronary imaging. For example, molecular imaging combined with intravascular ultrasound and optical coherence tomography may identify features of plaque rupture and of vascular inflammation, such as local macrophage accumulation and microchannel formation, as previously shown by our group.6 Such a strategy would require taking advantage of naturally occurring stressors, which are limited by unpredictability and confounding variables that are difficult to control for, unlike in experimentally induced MS. However, such stressors may be more valid and relevant to pathological mechanisms at play in the real world.
Hinterdobler et al. then extended their observations with a series of elegant experiments highlighting the central role of the endothelium (Graphical Abstract). They demonstrated that acute MS precipitated the release of local norepinephrine from peripheral sympathetic nerves innervating arterial walls. In turn, norepinephrine bound to α-adreno surface receptors on endothelial cells promoted up-regulation of cell adhesion molecules, such as intracellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), as well as release of the chemokines chemokine C–X–C motif ligand 1 (CXCL1) and chemokine C–C motif ligand 7 (CCL7). These effects led to leucocyte recruitment, vessel wall adherence, and vascular inflammation independent of haemodynamic changes. Further, these events were unique to local norepinephrine and were not observed with systemic epinephrine and corticosterone released from the adrenal medulla via sympathetic stimulation or the hypothalamic–pituitary–adrenal axis, respectively. In a secondary effect also involving endothelial cells, local macrophages and vascular smooth muscle cells were shown to release the chemokines tumour necrosis factor-α, interleukin (IL)-1β, and IL-6 in response to local norepinephrine thought to then signal to endothelial cells, further fuelling their activation. Thus, the authors highlight the pivotal role of the endothelium in cellular signalling that ultimately leads to vascular inflammation, plaque instability, and CVD events.
An important remaining question is whether the healthy endothelium behaves similarly to dysfunctional endothelium. The current study did not evaluate the structural integrity or functional health of the endothelial cells, an important factor in determining the cellular conditions required for MS to lead to CVD events. Indeed, given the location of the endothelium on the luminal surface of the vascular system, it is naturally most exposed to myriad insults. In clinical studies, a public speaking task7 and anger provocation8 were associated with an increase in circulating endothelial cell-derived microparticles derived from the membranes of apoptotic endothelial cells. Acute MS has also been shown to adversely influence endothelial cell function in animal studies,9 as well as in clinical studies in which experimental acute MS led to peripheral endothelial dysfunction (ED).10 Apical ballooning is, in fact, a human example of MS-induced transient myocardial injury, and is associated with both coronary and peripheral ED.11 Thus, the functional integrity of the endothelium may be the intermediary by which the pathological effects of acute MS are transduced, potentially through enhanced vascular permeability or other incompletely understood mechanisms that require exploration. ED often co-localizes with a vulnerable atherosclerotic plaque characterized by necrotic and lipid-rich cores and fibrous cap thinning,12 suggesting that ED could be the inciting pathological event, or at least a key biomarker.
Importantly, Hinterdobler et al. focused on mechanisms activated during acute MS. The relationship between chronic MS, atherosclerotic disease, and CVD events is less certain, with evidence suggesting a role for chronic stress in determining prognosis and outcomes in patients with pre-existing CVD, as opposed to causing CVD per se. 1 The authors have previously shown that chronic mild stress activates haematopoietic stem cells in the bone marrow, leading to increased leucocyte production.13 Similarly, using [18F]FDG PET/CT in conjunction with CT coronary angiography showed that increased amygdala activity was associated with greater bone marrow leucopoietic activity and coronary atherosclerosis,14 and functional magnetic resonance imaging revealed that individuals with heightened amygdala activation had a greater burden of subclinical atherosclerosis.15 Chronic stress may mediate acute events, but seems to play a distinct role in the manifestation of CVD compared with acute MS. Studies exploring the cellular mechanism underpinning this relationship would also be of great value and, given the tight coupling between inflammation, atherosclerosis, and ED, should similarly explore the role of the endothelium.
In their study, Hinterdobler et al. demonstrated that treatment with antibodies against the chemokines CXCL1 and CCL7 curtailed plaque leucocyte recruitment, thereby limiting vascular inflammation. Indeed, novel therapeutics targeting proinflammatory cytokines, such as canakinumab which acts on IL-1β, reduce rates of CVD events in patients with previous MI.16 Given our evolving knowledge of the neuroimmune axis at play during MS, such therapeutics could offer promise in this setting as well, and merit exploration in clinical trials. Further, ED may not only be a mechanistic key to the relationship between MS and CVD events, but might also constitute a therapeutic target representing accumulated vascular injury. Its detection could prompt CVD preventive efforts with behavioural modification, such as smoking cessation and physical activity, and the use of pharmacotherapy with drugs that improve ED, such as statins.17 Antidepressants such as selective serotonin re-uptake inhibitors reduce rates of CVD events in patients with recent acute coronary syndrome and depression,18 and their efficacy in preventing MS-induced CVD events should also be explored. The challenge, however, for controlled clinical trials investigating the utility of screening for and treating the effects of MS is first defining and provoking acute MS in a reliable and valid way and, perhaps more difficult, teasing apart its direct pathological effects on the neuroimmune and vascular systems.
Taken together, the study by Hinterdobler et al. helps to identify a number of key challenges and next steps in our attempt to learn more about this intriguing and relevant area, by shedding light on the central role of the endothelium as the effector cells of the neuroimmune response to MS.
Conflict of interest: none declared.
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.
References


{kind=link}