





Abstract
Despite advanced understanding of the biology of atherosclerosis, coronary heart disease remains the leading cause of death worldwide. Progress has been challenging as half of the individuals who suffer sudden cardiac death do not experience premonitory symptoms. Furthermore, it is well-recognized that also a plaque that does not cause a haemodynamically significant stenosis can trigger a sudden cardiac event, yet the majority of ruptured or eroded plaques remain clinically silent. In the past 30 years since the term ‘vulnerable plaque’ was introduced, there have been major advances in the understanding of plaque pathogenesis and pathophysiology, shifting from pursuing features of ‘vulnerability’ of a specific lesion to the more comprehensive goal of identifying patient ‘cardiovascular vulnerability’. It has been also recognized that aside a thin-capped, lipid-rich plaque associated with plaque rupture, acute coronary syndromes (ACS) are also caused by plaque erosion underlying between 25% and 60% of ACS nowadays, by calcified nodule or by functional coronary alterations. While there have been advances in preventive strategies and in pharmacotherapy, with improved agents to reduce cholesterol, thrombosis, and inflammation, events continue to occur in patients receiving optimal medical treatment. Although at present the positive predictive value of imaging precursors of the culprit plaques remains too low for clinical relevance, improving coronary plaque imaging may be instrumental in guiding pharmacotherapy intensity and could facilitate optimal allocation of novel, more aggressive, and costly treatment strategies. Recent technical and diagnostic advances justify continuation of interdisciplinary research efforts to improve cardiovascular prognosis by both systemic and ‘local’ diagnostics and therapies. The present state-of-the-art document aims to present and critically appraise the latest evidence, developments, and future perspectives in detection, prevention, and treatment of ‘high-risk’ plaques occurring in ‘vulnerable’ patients.
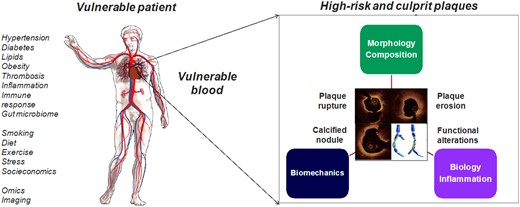
In the 1980s, Muller et al. studied coronary events triggered by morning awakening, anger, heavy exertion, and other stressors. The observation that a potential stressful trigger could be harmless on one day, yet lead to a cardiac event sometime later led to the concept of the ‘vulnerable plaque’, initially defined in 19891 as a plaque at increased risk of thrombosis (Supplementary material online, Table S1). Over the past three decades, the focus on ‘vulnerable plaque’1 , 2 (Supplementary material online, Figure S1) has evolved into a broader ‘vulnerable patient and plaque’ concept,3 , 4 as the current evidence suggests that clinically silent plaque ruptures may occur5 and that the coexistence of ‘vulnerable plaques’ in a ‘vulnerable patient’—including the prothrombotic milieu of circulating blood—is likely needed to generate acute coronary events (Take home figure) (Supplementary material online, Appendix—Evolution of the concepts of ‘vulnerable plaque’ and ‘vulnerable patient’). It has been also recognized that aside a thin-capped, lipid-rich plaque associated with culprit plaque rupture, acute coronary syndromes (ACS) are also caused by plaque erosion underlying between 25% and 60% of ACS nowadays,4 , 6 by calcified nodule or by functional coronary alterations.
High-risk and culprit plaques in ‘vulnerable patients’. The combination of ‘vulnerable patient’ phenotype and ‘high-risk plaque’ is likely required to produce an adverse cardiac event. Plaque rupture, plaque erosion, calcified nodule and functional coronary alterations represent pathophysiological mechanisms underlying acute coronary syndromes. Photography adapted from Partida et al. 6 and Gijsen et al. 7 (Creative Commons Licence CC-BY-NC).
Some small-to-moderate sample size longitudinal imaging studies have suggested that atherosclerotic plaques of differing maturity frequently co-exist and their morphology may change over time, being able to both gain and lose the features of vulnerability8–14; even up to three quarters of vulnerable plaques might evolve towards a more stable phenotype while on a high-intensity statin therapy.13
At present, the clinical consequences of detecting the features of plaque vulnerability remain limited as the positive predictive value of current imaging modalities for the prediction of major adverse cardiac events (MACE) is too low for clinical relevance. However, such limitations have prompted a number of technical advances including both diagnostics and analytics. In 2003, Patrick W. Serruys, Antonio Colombo, and Christodoulos Stefanadis founded an annual international meeting of medical scientists studying the detection and treatment of vulnerable plaques and patients2; the expert discussions that have occurred over the course of the past 15 years form the basis for this state-of-the-art document aiming to present and critically appraise the latest evidence, developments, and future perspectives in detection, prevention, and treatment of high-risk plaques occurring in ‘vulnerable’ patients.
As the majority of plaque ruptures or erosions are clinically silent, it is likely that coexistence of ‘high-risk plaques’ within ‘vulnerable patients’ is required to generate a clinical event/acute coronary syndrome.
Plaque rupture, plaque erosion, calcified nodule, and functional coronary alterations represent pathophysiological mechanisms underlying ACS.
Imaging of the high-risk/culprit coronary plaque
The search for the precursors of culprit plaque underlying acute cardiac events has been the subject of multiple investigations using different invasive and non-invasive imaging modalities.
Non-invasive imaging offers the potential as a screening tool to identify plaques with features of vulnerability, indicating patients at increased cardiovascular risk.15 Advanced computational modelling allows the use of increasing amount of radiographic data (radiomics)16 in clinical risk estimation, i.e. a new machine learning-based coronary computed tomography angiography (CCTA)-derived profiling of adverse fibrotic and microvascular perivascular adipose tissue remodelling has improved cardiac risk prediction using CCTA over and above the current state-of-the-art17 (Figure 1). The advances in non-invasive coronary imaging, including CCTA, magnetic resonance angiography, and positron emission tomography (Figure 2), have been recently reviewed in detail elsewhere.15

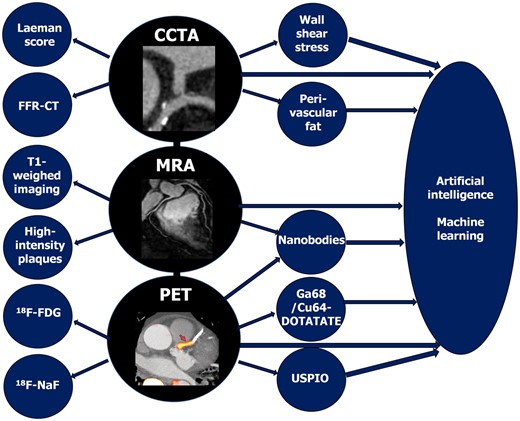
Schematic summary of advances in non-invasive imaging of coronary plaque. CCTA, coronary computed tomography angiography; FFR, fractional flow reserve; 18F-FDG, 18F-fluorodeoxyglucose; 18F-NaF, 18F-sodium fluoride; MRA, magnetic resonance angiography; PET, positron emission tomography; USPIO, ultrasmall superparamagnetic iron oxide. Photography adapted from Joshi et al. 20 and Chiribiri et al. 21
There are three intracoronary imaging modalities that have been tested for their ability to identify high-risk plaques and to evaluate culprit lesions: intravascular ultrasound (IVUS), intravascular optical coherence tomography (OCT), and near-infrared spectroscopy (NIRS) (Figure 3). They have also been utilized to identify a response to pharmacotherapy (Supplementary material online, Table S2).
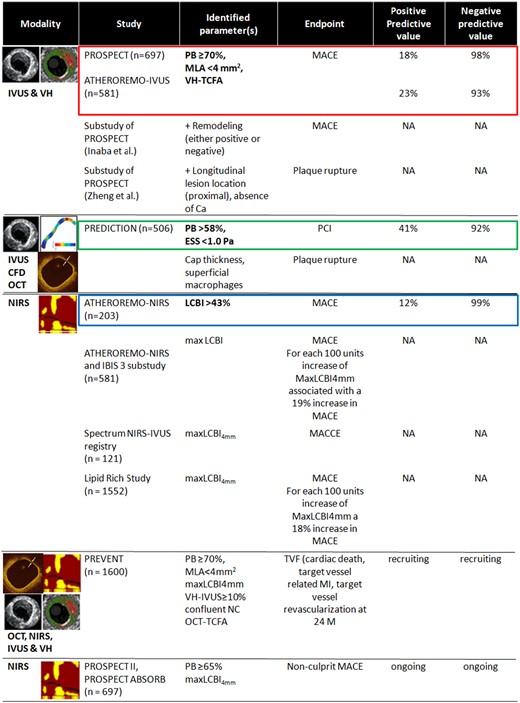
Intravascular imaging modalities in studies on high-risk plaques and vulnerable patients. CA, calcium; ESS, endothelial shear stress; IVUS, intravascular ultrasound; LCBI, lipid core burden index; MACE, major adverse cardiac events; MLA, minimum lumen area; NA, not available; NIRS, near-infrared spectroscopy; OCT, optical coherence tomography; PB, plaque burden; TCFA, thin-cap fibroatheroma; VH, virtual histology.
With IVUS, a large plaque burden, small lumen area, and positive remodelling have been demonstrated as strong markers of plaque ‘vulnerability’.22 Attenuated plaque or a necrotic core (especially with a non-detectable fibrous cap, <150 μm thick according to the resolution of the technique) by IVUS-virtual histology are also a sign of a plaque vulnerability.23 , 24 However, IVUS has only limited ability to identify the amount and distribution of lipid present in a plaque. NIRS, which has been combined with IVUS in a multimodal catheter, is quantitatively validated for the detection and characterization of lipid content of a plaque. The relatively low spatial resolution of IVUS limits its ability to assess the thickness of a fibrous cap over a lipid core; OCT, which has an ∼10-fold higher resolution, is superior for the detection of potentially vulnerable thin caps and remains the most reliable imaging modality to confirm plaque erosion. Finally, IVUS has no chemical sensitivity, which means that image contrast for inflammation or biological instability is absent. These may potentially be detected by local or systemic biomarkers, or molecular imaging.25
Near-infrared spectroscopy measures the lipid core burden based on the differential absorbance and scattering of NIR light at different wavelengths for different types of tissue. Several trials have demonstrated the value of NIRS in predicting MACE (Figure 3).26–28 Recently, the Lipid-Rich Plaque Study in 1562 patients demonstrated that non-flow-limiting plaques, which did not undergo percutaneous coronary intervention (PCI) but were shown to be lipid-rich by NIRS, were associated with increased MACE at both the patient and the plaque level 24 months following PCI of an index lesion.29
It should be noted, however, that the events triggered by occlusive thrombosis occurring over plaques without superficial lipid escape the accurate prediction with NIRS, a consideration particularly essential in the context of the increasing recognition of the role of plaque erosion as the cause of ACS.6 Currently, the PROSPECT II (ClinicalTrial.org Identifier: NCT02171065) and PREVENT (ClinicalTrial.org Identifier: NCT02316886) trials are underway, aiming to further define the value of NIRS-IVUS in prediction of coronary events; these studies are also evaluating the ability of IVUS, OCT, and NIRS to guide localized treatment (stenting) of plaques identified by imaging as having a high risk of becoming a future culprit lesion.
At present, the clinical consequences of detecting the ‘high-risk’ plaque features remain limited given that the positive predictive value of available invasive imaging modalities for the prediction of adverse events is too low for clinical relevance. However, such limitations have prompted a number of further technical advances.
Algorithms, using the gold standard of plaque burden determined by IVUS, are under development to improve the detection of plaque burden by OCT.30 Recent developments in IVUS and OCT, including higher resolution IVUS systems operating at 60 MHz, electrocardiography-gated, and Heartbeat OCT, provide a higher longitudinal sampling rate without artefact due to cardiac motion (Supplementary material online, Appendix—Developments in intravascular imaging).
Hybrid intravascular imaging catheter-based systems have recently been introduced to allow simultaneous acquisition of data by different modalities with complementary strengths31 (Supplementary material online, Appendix—Hybrid intravascular imaging catheters). The combined OCT-NIRS, IVUS-OCT/OFDI hybrid catheter systems, plus other novel approaches using near-infrared fluorescence (NIRF) molecular imaging, intravascular photoacoustics (IVPA), time-resolved fluorescence spectroscopy, and fluorescence lifetime imaging systems currently under development are summarized in Figure 4. Molecular imaging with NIRF measures inflammation using a ProSense marker and detects indocyanine green-illuminated plaque endothelial barrier function impairment.25 Hybrid intracoronary NIRF-OCT and NIRF-IVUS catheters, anticipated by 2020, may have a value in improving risk stratification in patients with established coronary artery disease (CAD).25 Fluorescence lifetime imaging, based on measurement of the fluorescence emission decay time after molecules have been excited with pulsed light. It can reportedly identify compositional characteristics of superficial plaque.32 Intravascular photoacoustics, a natural extension of IVUS that adds depth-resolved tissue to the IVUS images, relies on the analysis of the sound produced by the thermal expansion of tissues after irradiation with pulsed light. The photoacoustic response of each plaque depends on the absorption characteristics of its components, which permits IVPA to provide information about plaque composition. A study in an atherosclerotic animal model is ongoing.33
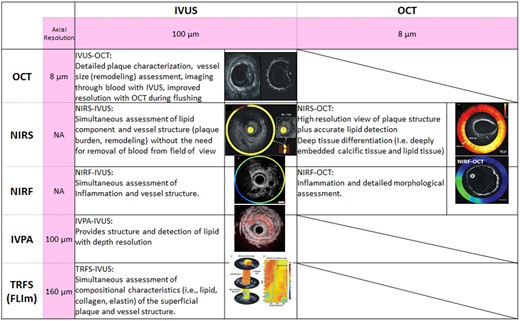
Summary of potential advantages of hybrid catheters. FLIm, fluorescence lifetime imaging; IVPA, intravascular photoacoustics; IVUS, intravascular ultrasound; NA, not available; NIRF, near-infrared fluorescence; NIRS, near-infrared spectroscopy; OCT, optical coherence tomography; TRFS; time-resolved fluorescence spectroscopy.
Can the assessment of local haemodynamic forces improve the identification of vulnerable plaques and patients?
Although the anatomic and chemical features of potentially vulnerable plaques have been extensively studied, the majority of these high-risk plaques remain quiescent and do not cause MACE.24 , 29 , 34 , 35 The low positive predictive value of current plaque characterization methods indicates that additional information is needed to identify vulnerable plaques in a manner that has clinical utility. There is increasing evidence that haemodynamic-associated biomechanical forces increase plaque vulnerability and may lead to destabilization causing a clinical event.35–38 The most extensively studied biomechanical forces are endothelial shear stress (ESS), plaque structural stress, and axial plaque stress.7 , 35 , 39 , 40 In addition, a fluid–structure interaction model is being developed to define pulsatility as a determinant of the risk of plaque rupture (Supplementary material online, Appendix—Impact of local haemodynamic forces on plaque biology). Although larger clinical studies and more data are required, initial reports suggest that inclusion of biomechanical indices in prediction models may increase the accuracy of predicting risk of plaque erosion41 , 42—a pathophysiological mechanism recognized as underlying a substantial proportion of acute coronary events.6
Prediction of plaque progression and vulnerability improves when high plaque burden coexists with areas of both low and high wall shear stress.35 The presence of low wall shear stress was associated with the development of non-culprit MACE in the post hoc computational fluid dynamics (CFD) analysis of patients enrolled in the PROSPECT study: baseline wall shear stress in patients with non-culprit plaques leading to MACE was lower than in patients who did not develop MACE during a median of 3.4 years of follow-up.36 It has been also shown that high wall shear stress in the upstream part of a plaque may contribute to the prognostic value of fractional flow reserve to predict myocardial infarction.37
Although CFD is a powerful research tool, such analysis requires numerous assumptions and procedures, including three-dimensional reconstruction of the vessel, meshing, and solving the Navier–Stokes equation. The principles of appropriate ESS computation and its potential clinical implications have been described in detail elsewhere.7
Non-invasive imaging offers the potential as a screening tool to identify patients at increased cardiovascular risk, while invasive imaging may further risk stratify patients who have presented with symptomatic CAD.
It is likely that plaque biomechanical forces act in a synergistic manner with anatomic and biochemical vulnerable plaque features to increase plaque vulnerability, promote destabilization, and produce clinical events.
New imaging modalities, multimodality catheters, and hybrid approaches, in conjunction with CFD processing are under development to improve the accuracy of predicting clinical events.
Prevention and treatment of culprit plaques and vulnerable patients
A systemic intervention with pharmacotherapy remains the key intervention in prevention of developing culprit lesions in vulnerable patients (Supplementary material online, Figure S2). The role of aspirin in primary prevention—that has long been advocated to mitigate the prothrombotic milieu of circulating blood in case the culprit lesion occurs—has been reappraised by three large randomized clinical trials showing minimal benefits and consistent bleeding risks.43 Thus, beyond diet maintenance, exercise, and smoking cessation, effective hypolipidaemic regimens rather than prophylactic aspirin are recommended for primary prevention.44
Improving coronary plaque imaging may be instrumental in guiding pharmacotherapy—its intensity and duration—and could facilitate optimal allocation of (such) novel, more ‘aggressive’, and costly treatment strategies (i.e. new hypolipidaemic or anti-inflammatory agents). While both pharmacological and mechanical intervention with PCI are well established in management of ACS caused by culprit plaque rupture, the concept of tailoring the treatment of ACS to its specific underlying mechanism, as assessed by intravascular imaging, is gaining an increasing interest. Specifically, the therapeutic approaches concentrating on antithrombotic agents rather than PCI in plaque erosion have been suggested, given the reported differences in vascular responses to PCI between plaque rupture and erosion, and the potential benefits of avoiding the risk of stent thrombosis, restenosis, and prolonged dual antiplatelet therapy.4 , 6
The single-centre EROSION study was the 1st proof-of-concept investigation that demonstrated the feasibility and initial safety of antithrombotic therapy without stenting in patients with OCT-confirmed culprit plaque erosion, further supporting evaluation of management schemes tailored to culprit plaque morphology.45
Over and above statins, therapeutic approaches to reduce LDL-C focused on a proprotein convertase subtilisin-kexin type 9 (PCSK 9) as a therapeutic target—a serine protease targeting low-density lipoprotein receptors for lysosomal degradation. Evolocumab and alirocumab, two human monoclonal antibodies directed against PCSK 9, administered on a background of statin therapy, have been shown to markedly decrease LDL-C levels and significantly reduce cardiovascular risk.46 With accumulating data supporting the efficacy of newer lipid-lowering agents in further reducing LDL-C,47 appropriate patients selection, therapy duration, and cost-effectiveness are gaining an increased interest. Although effective in stabilizing lipid-rich, thin-capped precursors of culprit plaque rupture, lipid-lowering agent might be less effective in preventing endothelial cell denudation and subsequent plaque erosion, that has been shown to frequently occur within lipid-poor regions of artery wall. A better understanding of the mechanisms that underlie superficial erosion is required to propose a more specific therapeutic approaches; innate immune activation of intimal endothelial cells (by engagement of the toll-like receptor 2), neutrophils, and neutrophil extracellular traps appears to contribute more prominently to thrombosis due to superficial erosion than in case of plaque rupture, pointing towards inflammatory pathways as promising new targets.48
Indeed, evidence of ongoing inflammatory processes in ruptured or eroded culprit plaques has led to trials assessing anti-inflammatory agents in the prevention of cardiovascular events.8 , 49 Canakinumab—a high-affinity fully human monoclonal anti-human interleukin-1β (IL-1β) antibody—demonstrated a beneficial effect on MACE in the CANTOS trial.46 , 50 Nevertheless, the translational aspects of these findings still need to be further elucidated, with a focus on individuals presenting high baseline inflammatory biomarker concentrations, subjects responding with a pronounced reduction of C-reactive protein or IL-6, and particular attention to side effects, such as infections. While a 1st trial using methotrexate yielded a null result,51 another anti-inflammatory compound—colchicine—has reduced the rates of MACE after a myocardial infarction in the recent COLCOT trial; further studies evaluating this compound are ongoing (LoDoCo2, CLEAR-SYNERGY, CONVINCE).
It should be noted, however, that the trials evaluating novel anti-inflammatory and lipid-lowering agents demonstrated a benefit of ∼15% reduction in mortality and adverse cardiovascular events, as compared to their control groups; there was still ∼85% residual risk of mortality and MACE that persisted despite these aggressive and innovative systemic therapies.46 , 50 If new local imaging and diagnostic strategies (anatomic, biochemical, and biomechanical) were able to identify the precursors of culprit plaques with greater prognostic discrimination, future pre-emptive local strategies targeting the remaining 85% risk could potentially provide incremental clinical benefit. With the improvements in stent design and secondary prevention drug therapy, PCI could recently reduce the risk of ‘hard’ clinical endpoints, including cardiovascular death or myocardial infarction, when performed in non-culprit lesions causing severe stenoses (≥80% by visual estimation or ≥60% by quantitative coronary angiography).52 If ongoing imaging and diagnostic efforts identify a truly high-risk features in jeopardy of destabilization and MACE in a plaque with <80% luminal obstruction, then pre-emptive strategies could be applied beyond the COMPLETE trial inclusion criteria,52 and potentially improve clinical outcomes more substantially. Nevertheless, to date there is no conclusive evidence demonstrating that sealing of high-risk plaques that are not significantly stenotic is clinically safe and improves prognosis.53
Atherosclerosis—as a systemic disease with focal manifestations—may require systemic pharmacologic therapy and local interventional treatment of advanced lesions.
Improving coronary plaque imaging may be instrumental in guiding pharmacotherapy, facilitating optimal allocation of novel, more aggressive, and costly treatment strategies and tailoring the treatment of ACS to a specific underlying mechanism (culprit plaque rupture, erosion, calcified nodule, or functional coronary alterations).
Conclusions and future perspectives
Despite significant advances in diagnostics, ranging from blood testing to genetics, imaging, haemodynamics, and ‘omics’, identification of patients and plaques at higher risk of adverse events remains limited. The positive predictive value of detecting the high-risk plaque characteristics remains too low for clinical relevance, and it is also still unclear which individual plaque features are most useful in predicting the ‘hard’ clinical endpoint of death and myocardial infarction. Nevertheless, improving imaging modalities supported by ongoing deep machine learning-based developments provide new avenues of precision medicine that are likely to translate into personalized preventive and therapeutic approaches to high-risk plaques occurring in ‘vulnerable’ patients. Further interdisciplinary research taking advantage of opportunities offered by both systemic and ‘local’ diagnostics and therapies is warranted.
This paper was guest edited by Filippo Crea.
Acknowledgements
The authors would like to thank the CVPath Institute, Inc. for help in the preparation of the figures for the manuscript and for the provided photographic materials.
Conflict of interest: M.T. acknowledges funding from the European Society of Cardiology as the laureate of the 2018 ESC Research and Training Grant Programme (Grant Nr: T-2018-19628). R.M. received research grant from Biosensors and SMT, outside the submitted work. R.Y.K. has a patent Antibody targeted imaging of oxidized LDL in atherosclerosis—Imperial College London. G.v.S. reports grants from Terumo Corp., grants from Shenzhen Vivolight, outside the submitted work; In addition, G.v.S. has a patent US9962127 and related international applications licenced to Terumo Corp., and a patent EP3256047 and related international applications issued; patents are held by institution. N.E.J.W. reports personal fees from PlaqueTec Ltd, outside the submitted work. A.M. reports an Institutional grant from Abbott Vascular, Boston Scientific. Consultant for Boston Scientific, Conavi Medical Inc., Philips. R.V. reports personal fees and non-financial support from CVPath Institute during the conduct of the work. R.V. reports institutional research support from R01 HL141425 Leducq Foundation Grant; 480 Biomedical; 4C Medical; 4Tech; Abbott; Accumedical; Amgen; Biosensors; Boston Scientific; Cardiac Implants; Celonova; Claret Medical; Concept Medical; Cook; CSI; DuNing, Inc.; Edwards LifeSciences; Emboline; Endotronix; Envision Scientific; Lutonix/Bard; Gateway; Lifetech; Limflo; MedAlliance; Medtronic; Mercator; Merill; Microport Medical; Microvention; Mitraalign; Mitra assist; NAMSA; Nanova; Neovasc; NIPRO; Novogate; Occulotech; OrbusNeich Medical; Phenox; Profusa; Protembis; Qool; Recor; Senseonics; Shockwave; Sinomed; Spectranetics; Surmodics; Symic; Vesper; W.L. Gore; Xeltis and received honoraria from Abbott Vascular; Biosensors; Boston Scientific; Celonova; Cook Medical; Cordis; CSI; Lutonix Bard; Medtronic; OrbusNeich Medical; CeloNova; SINO Medical Technology; ReCore; Terumo Corporation; W. L. Gore; Spectranetics; and is a consultant Abbott Vascular; Boston Scientific; Celonova; Cook Medical; Cordis; CSI; Edwards Lifescience; Lutonix Bard; Medtronic; OrbusNeich Medical; ReCore; Sinomededical Technology; Spectranetics; Surmodics; Terumo Corporation; W. L. Gore; Xeltis, outside the submitted work. W.K. reports personal fees from AstraZeneca, personal fees from Novartis, personal fees from DalCor, personal fees from Kowa, personal fees from Amgen, personal fees from Sanofi, grants and non-financial support from Roche Diagnostics, grants and non-financial support from Beckmann, grants and non-financial support from Singulex, grants and non-financial support from Abbott, outside the submitted work. G.W.S. reports personal fees from Terumo, personal fees from Amaranth, personal fees from Shockwave, personal fees from Valfix, personal fees from TherOx, personal fees from Reva, personal fees from Vascular Dynamics, personal fees from Robocath, personal fees from HeartFlow, personal fees from Gore, personal fees from Ablative Solutions, personal fees from Matrizyme, personal fees from Miracor, personal fees from Neovasc, personal fees from V-wave, personal fees from Abiomed, personal fees from Claret, personal fees from Sirtex, personal fees and other from Ancora, personal fees and other from Qool Therapeutics, other from Cagent, other from Applied Therapeutics, other from Biostar family of funds, other from MedFocus family of funds, personal fees and other from SpectraWave, personal fees from MAIA Pharmaceuticals, other from Orchestra Biomed, outside the submitted work; and Columbia University receives royalties from Abbott for sale of the MitraClip. J.E.M. reports personal fees and equity in SpectraWAVE, Inc., a company working to create an OCT-NIRS coronary catheter. W.W. has received speaker fees from Biotronik and MicroPort; he is a scientific advisor for Rede Optimus Research and co-founder of Argonauts, an innovation accelerator. P.W.S. reports personal fees from Abbott Laboratories, personal fees from AstraZeneca, personal fees from Biotronik, personal fees from Cardialysis, personal fees from GLG Research, personal fees from Medtronic, personal fees from Sino Medical Sciences Technology, personal fees from Société Europa Digital Publishing, personal fees from Stentys France, personal fees from Svelte Medical Systems, personal fees from Philips/Volcano, personal fees from St. Jude Medical, personal fees from Qualimed, personal fees from Xeltis, outside the submitted work. All other authors declared no conflict of interest.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}