





Abstract
Acute heart failure (HF) and in particular, cardiogenic shock are associated with high morbidity and mortality. A therapeutic dilemma is that the use of positive inotropic agents, such as catecholamines or phosphodiesterase-inhibitors, is associated with increased mortality. Newer drugs, such as levosimendan or omecamtiv mecarbil, target sarcomeres to improve systolic function putatively without elevating intracellular Ca2+. Although meta-analyses of smaller trials suggested that levosimendan is associated with a better outcome than dobutamine, larger comparative trials failed to confirm this observation. For omecamtiv mecarbil, Phase II clinical trials suggest a favourable haemodynamic profile in patients with acute and chronic HF, and a Phase III morbidity/mortality trial in patients with chronic HF has recently begun. Here, we review the pathophysiological basis of systolic dysfunction in patients with HF and the mechanisms through which different inotropic agents improve cardiac function. Since adenosine triphosphate and reactive oxygen species production in mitochondria are intimately linked to the processes of excitation–contraction coupling, we also discuss the impact of inotropic agents on mitochondrial bioenergetics and redox regulation. Therefore, this position paper should help identify novel targets for treatments that could not only safely improve systolic and diastolic function acutely, but potentially also myocardial structure and function over a longer-term.
Introduction
Heart failure (HF) is a leading cause for hospital admissions in developed countries worldwide,1 and its incidence is further increasing as average life expectancy rises. While the prognosis of patients with chronic HF has improved over the last decades, the prognosis of acute HF is still poor. Although only a small percentage of patients with acute HF present with cardiogenic shock (∼3%) and/or signs of hypoperfusion (∼15%), their in-hospital mortality is much higher than of patients with acute HF without these severe conditions.2 An important treatment option in these patients are inotropic agents to acutely increase cardiac output.3 However, since the use of most inotropic agents [in particular, catecholamines and phosphodiesterase (PDE)-inhibitors] is complicated by adverse short- and long-term effects, their use is not recommended in the absence of hypotension or hypoperfusion.3 On the other hand, the oldest inotropic agent (digitalis) can be safely applied to patients with chronic HF and improves morbidity.4 Here, we discuss the detailed mechanisms of inotropic agents to estimate which mechanisms of action may provide benefit in either acute or chronic HF.
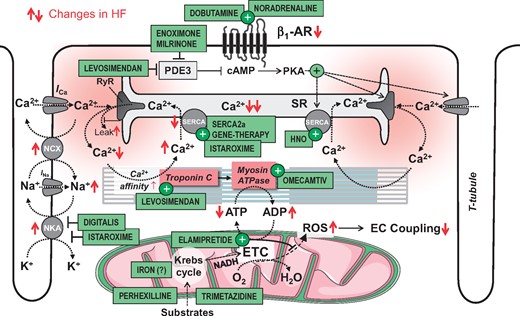
In patients with HF with reduced ejection fraction (HFrEF), the inability of the heart to eject sufficient blood for the needs of peripheral tissues is caused by defects of excitation–contraction (EC) coupling in cardiac myocytes (Figure 1).5 Traditionally, an acute increase in cardiac output is achieved with drugs that increase intracellular cyclic adenosine monophosphate (cAMP), such as catecholamines or (PDE) inhibitors (Figure 2). However, these agents increase myocardial oxygen (O2) consumption, predispose to life-threatening arrhythmias6 and activate signalling pathways of hypertrophy and cell death,7 , 8 which may explain why they are associated with adverse outcome.9 Consequently, recent developments aimed at increasing contractility without increasing cAMP or Ca2+ through Ca2+-sensitizing of myofilaments or myosin activation. Although meta-analyses with the Ca2+ sensitizer levosimendan suggest an overall benefit,10 , 11 larger comparative trials failed to show a survival benefit despite haemodynamic improvements.12–15 The myosin activator omecamtiv mecarbil has passed Phase II trials,16–20 and a Phase III trial was recently launched in patients with chronic HF (GALACTIC-HF; NCT0292932).
The physiology of excitation–contraction coupling and how this is altered in systolic heart failure. AR, adrenergic receptor; cAMP, cyclic adenosine monophosphate; ETC, electron transport chain; I Ca and I Na, Ca2+ and Na+ currents; NCX; Na+/Ca2+-exchanger; NKA, Na+/K+-ATPase; PDE, phosphodiesterase; PKA, protein kinase A; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; SERCA, SR Ca2+ ATPase; T-tubule, transversal tubule. Red arrows ( ↑↓) indicate the direction of change in heart failure.
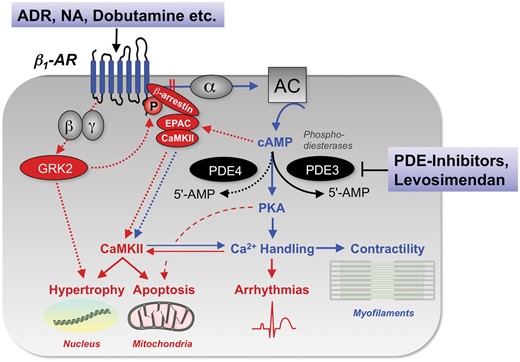
Signal transduction of β1-adrenergic stimulation in cardiac myocytes and its impact on inotropy, but also arrhythmias, hypertrophy, and apoptosis. 5’-AMP, 5’ adenosine monophosphate; AC, adenylyl cyclase; ADR, adrenaline; AR, adrenergic receptor; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cAMP, cyclic adenosine monophosphate; EPAC, exchange protein directly activated by cAMP; GRK2, G-protein coupled receptor kinase 2; NA, noradrenaline; PDE, phosphodiesterase; PKA, protein kinase A; α, β, γ, α-, β- and γ-subunits of the stimulatory G-protein.
Considering the poor outcome of patients in cardiogenic shock and/or signs of hypoperfusion2 and the mostly disappointing results of inotropic agents in patients with acute HF, novel strategies are urgently needed. To this end, the Committees on Translational Research and on Acute Heart Failure of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC) developed this position paper that addresses the following three key issues:
(1) Why have classical inotropic agents failed?
(2) Is direct targeting of sarcomere function therapeutically advantageous?
(3) Which novel concepts are promising?
To understand the mechanisms of traditional, current, and future medicines, it is essential to understand the physiology of EC coupling and its pathological alterations in HF. We will also discuss the bioenergetic consequences of inotropic interventions, since these may impact the long-term prognosis of HF patients. Finally, we will give recommendations for basic and clinical research directed at developing novel strategies for inotropic interventions in acute HF.
Physiology of excitation–contraction coupling
During each action potential, Ca2+ enters cardiac myocytes via L-type Ca2+ channels (LTCC), triggering even greater Ca2+ release from the Ca2+ stores of the cell, i.e. the sarcoplasmic reticulum (SR; Figure 1). This Ca2+ binds to troponin C, facilitating actin–myosin interaction that induces the contraction of the heart muscle. During diastole, Ca2+ diffuses away from troponin C, initiating relaxation. The Ca2+ that was released from the SR is taken back up by the SR Ca2+ ATPase (SERCA), whereas the amount of Ca2+ that entered the cell via LTCCs is exported by the Na+/Ca2+ exchanger (NCX).4
Cardiac contractility is increased by three principle mechanisms:
β-adrenergic stimulation,
the Frank–Starling mechanism and
the positive force-frequency relation (also known as ‘Bowditch-Treppe’).
Noradrenaline (NA) is released from sympathetic nerve endings in the myocardium and stimulates β1-adrenergic receptors (β1-ARs), which couple to the stimulatory G-protein (Gs) and activate the adenylyl cyclase (AC) to produce cAMP (Figures 1 and 2). Cyclic AMP activates protein kinase A (PKA) which (through phosphorylation) activates LTCCs, accelerates SERCA, increases SR Ca2+ release via ryanodine receptors (RyRs) and decreases myofilament Ca2+ affinity. Furthermore, cAMP activates the exchange protein directly activated by cAMP (Epac), further activating Ca2+/Calmodulin-dependent protein kinase II (CaMKII) which phosphorylates various Na+- and Ca2+-transporting proteins, mostly synergistic with PKA-mediated actions (Figure 2).21 The net result is an increase and acceleration of force generation and relaxation (positive inotropic and lusitropic effects), maintaining the refilling of the ventricles at elevated heart rates (positive chronotropic effect). In the human heart, the breakdown of cAMP is governed primarily by PDE3 and to a lesser extent by PDE4.22
Besides β-AR stimulation, the ‘Bowditch-Treppe’ increases cardiac contractility at elevated heart rates. This is explained by slower Ca2+-efflux (via the NCX) than -influx kinetics (via LTCCs), which results in intracellular accumulation of Ca2+ at higher heart rates that is sequestered into the SR by SERCA, from where it is released in greater amounts on the ensuing beat. The Frank–Starling mechanism is caused by a length-dependent increase of the Ca2+ affinity of the myofilaments, resulting in stronger contraction at unchanged cytosolic Ca2+ concentrations ([Ca2+]c).
Pathophysiological changes of excitation–contraction coupling in heart failure
The central deficit of EC coupling in myocytes from failing hearts is a decreased Ca2+ load of the SR. This is primarily the result of reduced SERCA expression and activity and a Ca2+ leak from the SR via RyRs, reducing systolic SR Ca2+ release and thereby the activator Ca2+ at the myofilaments (Figure 1).5 The reduced rate of SERCA-mediated Ca2+ re-uptake into the SR also slows relaxation. Furthermore, the cytosolic Na+ concentration ([Na+]i) is elevated in failing cardiac myocytes through changes in the ‘late Na+ current’, Na+/H+ exchanger (NHE) and Na+/K+-ATPase (NKA) activities.23 While this facilitates Ca2+-influx via the reverse mode of the NCX during the action potential,5 partly compensating for decreased systolic SR Ca2+ release,24 relaxation is further slowed by hampering diastolic Ca2+ extrusion via the forward mode NCX. This is particularly problematic at higher heart rates, when diastole progressively shortens. Consequently, the normally positive force–frequency relationship is blunted or even negative in failing human hearts, mediated by a variable combination of elevated diastolic [Ca2+]c and tension as well as decreased SR Ca2+ load and release.25 , 26 This decreases left ventricular ejection fraction (LVEF) and cardiac output at higher heart rates in vivo. 27 Finally, elevated diastolic [Ca2+]c and increased open probability of RyRs increases the probability of spontaneous SR Ca2+ release events, which (by subsequent Ca2+ extrusion via the electrogenic NCX) can induce delayed after-depolarizations, a well-defined trigger of ventricular arrhythmias.
In addition to the defects in ion handling and the ensuing inversion of the force–frequency relationship, continuous stimulation of cardiac β1-ARs through activation of the sympathetic nervous system desensitizes and downregulates β1-ARs (Figure 2),7 , 8 blunting their response to endogenous or exogenous catecholamines. Consequently, phosphorylation of several Ca2+ handling proteins is reduced. In failing hearts, the Ca2+ affinity of the myofilaments is increased as a result of decreased PKA-mediated phosphorylation of troponin I,28 , 29 although this issue is not fully settled yet.30
Energetic aspects
Excitation–contraction coupling requires high amounts of energy in the form of ATP, which is replenished by oxidative phosphorylation in mitochondria. During β-adrenergic stimulation, mitochondria take up Ca2+ to stimulate the Krebs cycle, which produces NADH as the main electron donor for ATP production at the respiratory chain (Figure 3).31 Mitochondrial function is impaired in HF, resulting in energetic deficit and oxidative stress (Figure 3).31–33 Mitochondrial dysfunction is linked to defects in EC coupling, since the Krebs cycle requires stimulation of its key enzymes by Ca2+, and decreased SR Ca2+ release hampers mitochondrial Ca2+ uptake via the uniporter (MCU). In addition, elevated [Na+]i accelerates mitochondrial Ca2+ efflux via the mitochondrial Na+/Ca2+ exchanger (NCLX; Figure 3). Impaired Krebs cycle activity limits NADH-dependent ATP production at the respiratory chain and provokes excess emission of reactive oxygen species (ROS) through depletion of the NADPH-dependent anti-oxidative capacity, causing oxidative stress.31 Reduced ATP production can limit the contractile reserve of the LV,34 and most Na+ and Ca2+ transporting mechanisms are sensitive to redox-dependent modifications.35 Thus, the tight interplay between EC coupling and mitochondrial energetics (Figure 3) can set in motion a vicious cycle of deteriorated ion handling, energetic deficit, and oxidative stress to aggravate systolic and diastolic dysfunction in HF.
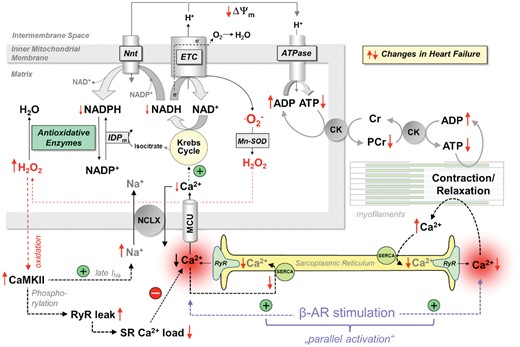
Interplay between EC coupling and mitochondrial energetics. Krebs cycle activity is controlled by Ca2+, and mitochondrial Ca2+ uptake is diminished in heart failure by changes in cytosolic Ca2+ and Na+ handling. This provokes an energetic deficit and oxidative stress, which further impairs EC coupling and aggravates systolic and diastolic function. AR, adrenergic receptor; ATPase, F1Fo-ATP synthase; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CK, creatine kinase; Cr, creatine; ETC, electron transport chain; IDPm, isocitrate dehydrogenase; late I Na, late Na+ current; MCU, mitochondrial Ca2+ uniporter; Mn-SOD, mitochondrial superoxide dismutase; NCLX, mitochondrial Na+/Ca2+-exchanger; Nnt, nicotinamide nucleotide transhydrogenase; PCr, phosphocreatine; RyR, ryanodine receptor; SERCA, SR Ca2+ ATPase. Red arrows ( ↑↓) indicate the direction of change in heart failure.
In patients with HF, iron deficiency (ID) predicts adverse outcome,36 while iron supplementation improves functional capacity and quality of life.37 In failing hearts, myocardial iron content is reduced and associated with reduced activity of Krebs cycle dehydrogenases and expression of anti-oxidative enzymes.38 In preclinical models, severe cardiac or skeletal muscle ID perturbs mitochondrial function and induces systemic metabolic derangements and cardiomyopathy.39 , 40 Accordingly, ID may further aggravate energy supply and demand mismatch and oxidative stress in HF. However, since plasma ID does not directly correlate with myocardial ID, these issues require further investigation (for more in-depth discussion see ref. 41).
‘Classical’ inotropic agents
Digitalis
Digitalis-derived cardiotropic glycosides (CTG) are the oldest inotropic drugs and increase [Na+]i in cardiomyocytes by inhibiting Na+ export via the NKA (Figure 4). [Na+]i accumulation hampers diastolic Ca2+ extrusion via the NCX and supports Ca2+ influx via the reverse mode NCX during systole, thereby increasing diastolic [Ca2+], Ca2+ transient amplitudes and consequently, inotropy (Figure 4). On the other hand, elevated [Na+]i accelerates mitochondrial Ca2+ efflux via the NCLX, reducing Ca2+-activation of the Krebs cycle and its regeneration of NADH and NADPH. Since NADPH is required for anti-oxidative enzymes to detoxify ROS (Figure 3), CTG-induced NADPH oxidation increases mitochondrial ROS emission and thereby arrhythmias (Figure 4).42 The pro-arrhythmic actions of CTG narrow its therapeutic range.43
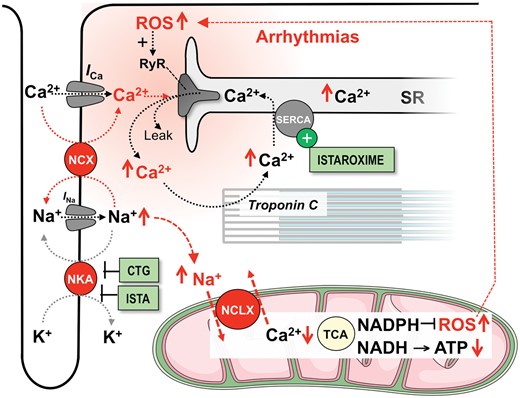
Mechanisms of action of cardiotonic glycosides (CTG) and istaroxime (ISTA). I Ca and I Na, Ca2+ and Na+ currents; NCLX, mitochondrial Na+/Ca2+-exchanger; NCX, Na+/Ca2+-exchanger; NKA, Na+/K+-ATPase; ROS, reactive oxygen species; RyR, ryanodine receptor; SERCA, SR Ca2+ ATPase; SR, sarcoplasmic reticulum; TCA, tricarboxylic acid (Krebs) cycle. Red arrows ( ↑↓) indicate the direction of change in response to CTG.
In patients with HFrEF, digoxin may be considered in symptomatic patients in sinus rhythm despite treatment with an ACE-inhibitor (or angiotensin receptor blocker), a β-blocker, and a mineralocorticoid antagonist to reduce the risk of hospitalizations (Class IIb, B3). However, the Digitalis Investigation Group (DIG) Trial4 was conducted before β-blockers became a mainstay of HF therapy, and digoxin did not improve all-cause mortality, but reduced hospitalization due to HF worsening. At the lower dosages used in current practice, digitalis may be preferentially a modulator of autonomic tone and less inotropic stimulator.44 The ongoing DIGIT-HF study prospectively investigates the role of digitoxin in patients with HFrEF already on current guideline-adherent therapy (http://digit-hf.de). In atrial fibrillation, clinical evidence is limited to small and observational studies, and there is an ongoing debate on potentially increased mortality.45 However, prescription bias in the retrospective analyses likely mimics the perceived digoxin driven mortality.46 According to the current HF Guidelines, an intravenous bolus of digoxin should be considered in digoxin-naïve-patients in New York Heart Association (NYHA) Class IV and rapid ventricular rate to slow heart rate (Class IIa, B).3
Catecholamines
The three endogenous catecholamines dopamine, adrenaline, and NA stimulate β1-ARs, and to variable degrees also α1- and β2-ARs and dopaminergic D1- and D2-receptors (Table 1).6 Thus, their haemodynamic actions are not limited to increasing cardiac contractility via β1-ARs (Figure 2). In fact, α1-AR-mediated vasoconstriction by adrenaline and NA (Table 1) renders them useful in patients with septic shock where vasodilation underlies hypotension. The haemodynamic profile of dopamine, the immediate precursor to NA in the synthetic pathway, is dominated by vasodilation at low concentrations at which binding to dopaminergic D1- and D2-receptors increases blood flow to the heart, brain, kidney, and various other organs. Its traditional use to increase renal blood flow, however, was discouraged after neutral effects in trials on patients with acute decompensated HF, with or without renal dysfunction.47–49 At higher doses, dopamine increases blood pressure by vasoconstriction via α1-ARs and positive inotropic and chronotropic effects via β-ARs, respectively.
Adrenergic affinities of endogenous and synthetic catecholamines and their effects on haemodynamics
| Drug | α1 | β1 | β2 | D1/D2-R | SVR | SV | HR | Arrh. |
|---|---|---|---|---|---|---|---|---|
| Adrenaline | ++++ | +++ | ++++ | 0 | ++ | ++++ | +++ | +++ |
| Noradrenaline | ++++ | +++ | + | 0 | ++++ | +++ | + | +++ |
| Dopamine | +++ | ++ | +++ | ++++ | +++ | ++++ | ++ | |
| Dobutamine | ++ | +++ | + | 0 | −/0/+ | ++++ | + | + |
| Isoproterenol | 0 | ++++ | ++++ | 0 | −−− | +++ | ++++ | +++ |
| Phenylephrine | ++++ | + | + | 0 | ++++ | −/0 | (−) |
| Drug | α1 | β1 | β2 | D1/D2-R | SVR | SV | HR | Arrh. |
|---|---|---|---|---|---|---|---|---|
| Adrenaline | ++++ | +++ | ++++ | 0 | ++ | ++++ | +++ | +++ |
| Noradrenaline | ++++ | +++ | + | 0 | ++++ | +++ | + | +++ |
| Dopamine | +++ | ++ | +++ | ++++ | +++ | ++++ | ++ | |
| Dobutamine | ++ | +++ | + | 0 | −/0/+ | ++++ | + | + |
| Isoproterenol | 0 | ++++ | ++++ | 0 | −−− | +++ | ++++ | +++ |
| Phenylephrine | ++++ | + | + | 0 | ++++ | −/0 | (−) |
− to −−−, increasing degrees of negative effects; + to ++++, increasing degrees of positive effects; 0, neutral effect; Arrh., arrhythmias; HR, heart rate; SV, stroke volume; SVR, systemic vascular resistance.
Adrenergic affinities of endogenous and synthetic catecholamines and their effects on haemodynamics
| Drug | α1 | β1 | β2 | D1/D2-R | SVR | SV | HR | Arrh. |
|---|---|---|---|---|---|---|---|---|
| Adrenaline | ++++ | +++ | ++++ | 0 | ++ | ++++ | +++ | +++ |
| Noradrenaline | ++++ | +++ | + | 0 | ++++ | +++ | + | +++ |
| Dopamine | +++ | ++ | +++ | ++++ | +++ | ++++ | ++ | |
| Dobutamine | ++ | +++ | + | 0 | −/0/+ | ++++ | + | + |
| Isoproterenol | 0 | ++++ | ++++ | 0 | −−− | +++ | ++++ | +++ |
| Phenylephrine | ++++ | + | + | 0 | ++++ | −/0 | (−) |
| Drug | α1 | β1 | β2 | D1/D2-R | SVR | SV | HR | Arrh. |
|---|---|---|---|---|---|---|---|---|
| Adrenaline | ++++ | +++ | ++++ | 0 | ++ | ++++ | +++ | +++ |
| Noradrenaline | ++++ | +++ | + | 0 | ++++ | +++ | + | +++ |
| Dopamine | +++ | ++ | +++ | ++++ | +++ | ++++ | ++ | |
| Dobutamine | ++ | +++ | + | 0 | −/0/+ | ++++ | + | + |
| Isoproterenol | 0 | ++++ | ++++ | 0 | −−− | +++ | ++++ | +++ |
| Phenylephrine | ++++ | + | + | 0 | ++++ | −/0 | (−) |
− to −−−, increasing degrees of negative effects; + to ++++, increasing degrees of positive effects; 0, neutral effect; Arrh., arrhythmias; HR, heart rate; SV, stroke volume; SVR, systemic vascular resistance.
Since in patients with cardiogenic shock, neuroendocrine activation induces vasoconstriction and tachycardia, the ideal drug should be positive inotropic without further increasing systemic vascular resistance (SVR). Dobutamine fulfils these requirements.50 It is a full agonist at β1-ARs, inducing a positive inotropic effect with similar efficacy as isoproterenol, a synthetic β1- and β2-AR agonist without any α-AR agonism (Table 1). The affinity of dobutamine for β2-AR is ∼10-fold lower than for β1-ARs and in particular, its agonist efficacy at β2-ARs and α1-ARs much weaker than at β1-ARs.51 Through its dominating inotropic effect and mutually offsetting vascular effects of α1- and β2-AR agonism, the decrease of SVR at intermediate and higher doses is mediated by reflex withdrawal of the endogenous sympathetic tone.52 For any given increase in cardiac contractility, the increase of heart rate and blood pressure is lower with dobutamine than with dopamine or NA, further reflecting dobutamine’s selectivity for β1- over β2- and α1-ARs.50 However, this favourable haemodynamic profile comes at the cost of elevated myocardial O2 consumption50 , 52 and arrhythmias.
Noradrenaline may be considered in patients who have cardiogenic shock despite treatment with another inotrope to increase blood pressure and vital organ perfusion.3 The combination of adrenaline with dobutamine, however, portends a particular risk for adverse outcome.53 In a recent meta-analysis, adrenaline was associated with a three-fold increase in mortality,54 and in patients with cardiogenic shock after acute myocardial infarction, refractory shock was five-fold more frequent with adrenaline than with NA.55 For a similar effect on blood pressure, adrenaline (but not NA) increased heart rate (due to its strong β2-AR activation; Table 1) and myocardial oxygen consumption (derived from the cardiac double product), increasing lactate as a sign of metabolic compromise.55 These data underscore that adrenaline should be avoided in patients with cardiogenic shock.
In patients with HF, elevated plasma NA levels predict adverse outcome.56 Chronic β-AR stimulation desensitizes and downregulates β-ARs via PKA, GRK2 (also known as β-ARK1), and β-arrestin.7 , 8 , 57 Furthermore, β-arrestin activates CaMKII which sustains contractility despite desensitization of β-ARs from PKA-mediated inotropy (Figure 2).21 CaMKII activation, however, is a major driver of cardiac arrhythmias.58 The net functional consequence of all these processes is a decreased responsiveness of the human failing heart to β-AR stimulation. Therefore, despite its favourable short-term haemodynamic profile that improves symptoms,9 dobutamine treatment is associated with tolerance,59 arrhythmias, and mortality, respectively.60 , 61
Phosphodiesterase-inhibitors
To overcome desensitization and down-regulation of cardiac β-ARs as well as their blockade through β-blockers, PDE-inhibitors were developed. In human failing myocardium, inhibition of PDE3, but not PDE4 potentiates β-AR-mediated positive inotropic effects.62 Conversely, in human atrial myocardium, inhibition of PDE4 potentiates arrhythmias induced by both β1- and β2-AR stimulation, while PDE3 inhibition only potentiates β1-AR-induced arrhythmias.63 Phosphodiesterase-inhibitors also decrease SVR through cAMP-mediated vasodilation, which is beneficial in patients with acute HF and high SVR, but limits its application in patients with cardiogenic shock due to reductions in blood pressure. In patients with HF treated with β-blockers, the efficacy and potency of PDE-inhibitors is maintained, while the effects of dobutamine are blunted.64
Why have classical inotropes failed?
Currently, dobutamine, dopamine, and PDE-inhibitors are recommended in patients with hypotension (SBP <90 mmHg) and/or signs/symptoms of hypoperfusion despite adequate filling status to increase cardiac output and blood pressure and to improve peripheral perfusion and maintain end-organ function (IIb, C).3 However, while in analyses that evaluated cAMP-dependent inotropes in general, their short-term use yielded neutral effects, longer-term use was associated with adverse outcome in patients hospitalized for acute HF despite improved quality of life.60 , 65 , 66 Therefore, inotropic agents are explicitly restricted to patients that fulfil the above mentioned criteria and are not recommended for any other patients (Class IIIA).3 The adverse long-term effects may be related to PKA- and CaMKII-induced maladaptive cardiac remodelling through inducing hypertrophy, apoptosis, and fibrosis (Figure 2).7 , 8 In particular, activation of β1-ARs induces apoptosis,67 which is an important mechanism for LV remodelling and dysfunction in HF.68 , 69 Furthermore, β-adrenergic activation alters myocardial substrate utilization and thereby, may trigger energetic deficit and oxidative stress.61 These data indicate that despite their favourable acute haemodynamic profile, the use of adrenergic agonists and PDE-inhibitors leads to adverse outcome by triggering maladaptive cardiac remodelling and arrhythmias, while vice versa, antagonizing β1-ARs can reverse remodelling, improve LV function, and prolong survival.7 Therefore, a new generation of inotropes had to be developed to avoid activation of adrenergic pathways and increase contractility without raising Ca2+, which is pro-arrhythmic and causes higher energy consumption through activation of Ca2+ transporting systems.
Treatments targeting sarcomeres
Ca2+ sensitizers
Ca2+ sensitizers shift the relationship between [Ca2+]c and force development of sarcomeres (i.e. the pCa–force relationship) to the left, increasing force at any given [Ca2+]c. They were developed in the early 1980s as cardiotonic agents with a number of theoretical advantages over catecholamines and PDE-inhibitors:
Ca2+ sensitizers should neither increase trans-sarcolemmal influx of Ca2+ nor alter SR Ca2+ fluxes during systole or diastole. This should be less pro-arrhythmic and less energy consuming.
Ca2+ sensitizers should not affect heart rate or blood pressure if a compound was selective for cardiac myofilaments, which may have positive effects on energetics.
Ca2+ sensitizers should be independent of the desensitized β-AR/cAMP system in HF and should themselves not induce tolerance.
Examples of this class of drugs are EMD-57033, CGP-48506, pimobendan, and levosimendan. The mechanisms of Ca2+ sensitization differ between Ca2+ sensitizers. While levosimendan and pimobendan increase the affinity of troponin C to bind Ca2+,70–72 CGP-48506 acts downstream of troponin C, and EMD-57033 affects the actin–myosin interaction by direct binding to the myosin motor domain.73 , 74 Independent of the mechanism, the shift of the pCa-force curve to the left increases systolic force generation for any given [Ca2+]c, but on the other hand impedes relaxation following the decrease in [Ca2+]c. The slowing of relaxation is an inherent property of pure Ca2+ sensitizers and may be the reason why most pharmaceutical companies stopped their development. In this context, mutations in sarcomeric proteins that cause hypertrophic cardiomyopathy (HCM) commonly increase myofilament Ca2+ sensitivity as a unifying disease mechanism.75 , 76 Moreover, both HCM mutations and drugs that increase Ca2+ sensitivity are arrhythmogenic, presumably by providing a sink for Ca2+ that is released during diastole, causing depolarisations via the electrogenic NCX.77 , 78
Levosimendan
In contrast to CGP-48506, levosimendan does not prolong relaxation time or compromise diastolic relaxation. Thus, it does not have the same profile as pure Ca2+ sensitizers. The most likely reason is that levosimendan is not only a Ca2+ sensitizer binding to troponin C,71 but also a potent and selective PDE3-inhibitor with an IC50 in the nanomolar range, but ∼1000-fold lower affinity for PDE4.79 , 80 Accordingly, levosimendan increases cAMP with similar potency as it increases force.81 Furthermore, its positive inotropic effect is abolished by the muscarinic receptor agonist carbachol, which acts through inhibiting cAMP generation. Also in human myocardium, the inotropic effects of levosimendan require β-adrenergic pre-stimulation and/or elevations of [Ca2+]c and can be prevented by PDE3-, but not PDE4-inhibition.82–84
Levosimendan’s clinical activity during long-term treatment is mainly governed by its active metabolite OR-1896, which has a much longer half-life (81 vs. 1 h85). OR-1896 stimulated contractile force with a roughly similar potency as levosimendan, had a 4.5-fold lower potency as a Ca2+ sensitizer and a 38-fold lower potency as a PDE3 inhibitor.80 Yet, even the inotropic effect of OR-1896 is sensitive to carbachol and therefore likely mediated by PDE3 inhibition.86 Therefore, also for OR-1896, a combination of PDE3-inhibition with Ca2+ sensitization is the mechanism that is responsible for positive inotropy.
A common alternative explanation why levosimendan does not prolong relaxation (other than through PDE3-inhibition) is that levosimendan’s binding to troponin C is Ca2+-dependent. However, it is currently unclear—if not rather unlikely—whether levosimendan can bind and unbind troponin C on a beat-to-beat basis in a millisecond and micromolar range, as would be required to explain the lack of relaxation prolongation by this mechanism (see the Supplementary material online for a detailed discussion of this issue). Besides its effects on EC coupling, levosimendan also activates glibenclamide-sensitive sarcolemmal ATP-dependent K+-currents (I KATP), which may add to its vasodilating activity and potentially provide cardioprotective effects through activation of mitochondrial I KATP (see Supplementary material online for a more detailed discussion).
These data indicate that for levosimendan, PDE3 inhibition synergizes with Ca2+ sensitization for its inotropic action (Figure 5), which may be particularly relevant to human failing myocardium in which PDE3 plays the dominant role for controlling intracellular cAMP.62 From this it can be predicted that the more β-ARs are pre-activated by endogenous or exogenous catecholamines, the more pronounced is the inotropic effect of levosimendan, and the more this effect is mediated by PDE3-inhibition rather than Ca2+ sensitization. Conversely, at low β-AR pre-activation (such as during pharmacological β-blockade), the Ca2+ sensitization effect of levosimendan may become more important for inotropy. In this context, it is interesting to observe that in the SURVIVE trial, patients with (but not without) β-blocker pre-treatment had improved short-term survival with levosimendan compared to dobutamine.87 , 88 Conversely, in patients with septic shock who were all co-treated with catecholamines, levosimendan was associated with higher rates of supraventricular tachycardia and a numerical, but non-significant increase in mortality (hazard ratio 1.24, P = 0.17).89 Therefore, although the clinical evidence for these considerations is limited, levosimendan's PDE3-inhibitory effect may be potentiated through pre-activation of β-ARs by endogenous or exogenous catecholamines also under in vivo conditions, and such potentiated adrenergic signalling may potentially contribute to arrhythmias and adverse consequences for cardiomyocyte biology (Figure 2).

Mode of action of levosimendan and its active metabolite OR-1896. Both Ca2+-sensitization and PDE3-inhibition at nanomolar concentrations (nM) contribute to their inotropic and lusitropic effects. Activation of mitochondrial KATP (mitoKATP) channels at micromolar concentrations (µM) may provide protection against ischaemia/reperfusion. AR, adrenergic receptor; cAMP, cyclic adenosine monophosphate; ETC, electron transport chain; I Ca and I Na, Ca2+ and Na+ currents; NCX, Na+/Ca2+-exchanger; NKA, Na+/K+-ATPase; PDE, phosphodiesterase; PKA, protein kinase A; RyR, ryanodine receptor; SERCA, SR Ca2+ ATPase; SR, sarcoplasmic reticulum; T-tubule, transversal tubule. Red arrows ( ↑↓) indicate the direction of change in heart failure, while green arrows ( ↑↓) indicate the direction induced by levosimendan.
Clinical trials
Several clinical trials tested the effects of levosimendan in patients with HF, comparing it to either placebo or dobutamine. In the LIDO trial, levosimendan improved haemodynamics more effectively than dobutamine and was associated with lower mortality than dobutamine after 180 days.90 In the SURVIVE trial, however, mortality after 180 days (the primary endpoint) was not different between dobutamine and levosimendan despite a more favourable haemodynamic profile (BNP reduction) in the first 5 days after randomization.12 As mentioned above, pre-treatment with a β-blocker was associated with improved short-term outcome at day 5 in a post hoc analysis.87
In the REVIVE trial,13 levosimendan (compared to placebo) was associated with more frequent hypotension and cardiac arrhythmias during the infusion period, and a numerical (but insignificant) risk of death despite improved symptoms and reduced plasma BNP levels. In a meta-analysis on 5480 patients in 45 randomized clinical trials, however, levosimendan was associated with a 20% relative risk reduction of mortality, and this reduction was confirmed in studies with placebo (−18%; P < 0.05) or dobutamine as comparator (−32%; P < 0.005).10 These trends were confirmed by another meta-analysis.11 However, in these meta-analyses, trials on patients with acute cardiac events were combined with trials on patients undergoing elective cardiac surgery. Furthermore, in the recent CHEETAH14 and LEVO-CTS15 trials, levosimendan did not improve outcome of patients with systolic HF undergoing cardiac surgery, although the use of inotropes 24 h after surgery was reduced with levosimendan in LEVO-CTS.15
Several smaller trials evaluated the usefulness of repeated doses of levosimendan for patients with advanced HF in outpatient settings. Post hoc and meta-analyses of these mostly underpowered trials suggest that levosimendan may have favourable effects on haemodynamics, symptoms, rehospitalization, and biomarkers.91 In the recent LION-HEART study on 69 patients with advanced HF, biweekly infusions of levosimendan for 12 weeks reduced NT-proBNP, improved quality of life and reduced hospitalization without adverse effects.92 Therefore, the initiation of a larger trial to test this treatment strategy for advanced HF patients is warranted.
Taken together, the principle of Ca2+ sensitization alone was no breakthrough in the treatment of the common forms of HF, because it is associated with worsening of diastolic relaxation, which is already compromised in HF in the first place. The ancillary PDE-inhibitory effect of levosimendan improves its haemodynamic profile compared to other, more pure Ca2+ sensitizers, although this may come at the cost of cAMP-related side effects (e.g. arrhythmias). So far, there is no clear evidence that levosimendan improves survival compared to placebo or a comparator drug.
Based on its clinical profile, the current HF Guidelines make the following recommendations for the use of levosimendan:
Short-term intravenous infusion of levosimendan may be considered in patients with hypotension (SBP <90 mmHg) and/or signs/symptoms of hypoperfusion despite adequate filling status to increase cardiac output and improve peripheral perfusion and maintain end-organ function (similar to dobutamine, dopamine, and PDE-inhibitors; IIb, C).3
When mean arterial pressure needs pharmacological support, a vasopressor (preferably NA) may be used in combination with levosimendan.3
An intravenous infusion of levosimendan (or a PDE inhibitor) may be considered to reverse the effect of β-blockade if β-blockade is thought to be contributing to hypotension with subsequent hypoperfusion (IIb, C).3
Levosimendan is not recommended unless the patient is symptomatically hypotensive or hypoperfused because of safety concerns (IIIA).3
Omecamtiv mecarbil
Omecamtiv mecarbil (OM) is a small-molecule, selective cardiac myosin activator whose therapeutic rationale and discovery were described elsewhere.93 , 94 Omecamtiv mecarbil binds to the catalytic domain of cardiac myosin, stabilizing the pre-powerstroke state,95 thus increasing the transition rate of myosin into the strongly actin-bound force-generating state (Figure 6A)96 and thus increasing cardiac contractility. The pharmacodynamic signature of OM is an increase in the systolic ejection time (SET). This is a consequence of the increase in the number of myosin heads interacting with actin filaments, facilitating a longer duration of systole, even as [Ca2+]c already decays. Omecamtiv mecarbil prolongs the time and increases the amplitude, but not the rate of cell shortening, and does not interfere with [Ca2+]c transients (Figure 6B).96
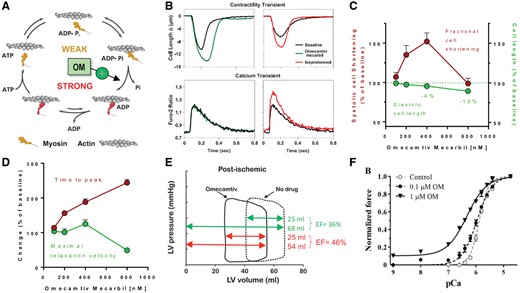
Mechanism of action and effects of omecamtiv mecarbil. (A) The mechanochemical cycle of myosin. Yellow indicates myosin weakly bound to actin, while red indicates the myosin strongly bound to actin. Omecamtiv mecarbil (OM) accelerates the transition rate of myosin into the strongly actin-bound force-generating state. (B) Representative tracings showing that OM (200 nM) increases the time and amplitude of myocyte shortening without any effect on the cytosolic Ca2+ transient. In contrast, the β-AR agonist isoproterenol increases myocyte shortening through increasing cytosolic Ca2+ transients. Fractional systolic sarcomere shortening and diastolic cell length (C) as well as time to peak and maximal relaxation velocity (D) in isolated rat cardiac myocytes in response to escalating concentrations of OM. (A–D) are from Malik et al. 96 with permission. (E) Impact of OM (20 min infusion at a dose that prolonged SET by 20%) on LV pressure-volume loops in a pig model of myocardial stunning (termed ‘post-ischaemic’ heart). The volumes indicate LV stroke volume and end-diastolic volume, of which EF is calculated. Taken from Bakkehaug et al. 99 with permission. (F) The impact of OM at 0.1 or 1 µM on normalized isometric force in response to increasing Ca2+ concentrations (decreasing pCa) in skinned rat cardiac myocytes. Taken from Nagy et al. 102 with permission.
A number of dose-finding studies with an intravenous formulation of OM were performed in a dog model of HF,97 healthy men17 and patients with acute19 and chronic HF,16 respectively. In the dog HF model, OM prolonged SET and increased stroke volume (SV; Table 2).96 , 97 Consequently, cardiac output increased, thereby decreasing SVR and heart rate, presumably through reducing endogenous sympathetic activation.96 , 97 Although OM might increase ATP turnover at the level of the sarcomere,96 this increase appears to be counterbalanced by the reduction in heart rate, SVR (reducing cardiac afterload) and end-diastolic volume (reducing myocardial wall stress), resulting in no significant change of cardiac O2 consumption despite the increase of cardiac output.96 , 97 Consequently, cardiac efficiency should improve.
Effects of omecamtiv mecarbil on haemodynamics in large animal models and clinical trials
| Study | Species, condition | Appl. | n | Time of OM admin. | OM Plasma Conc. (ng/mL) | SET | SV | HR | C. O. | SVR | LVEDP | LVEDV (LVEDD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shen | Dog MI/Pacing | i.v. | 6 | 15 min–72 h | ? | +20-30% | +44% | −15% | +22% | −15% | −17% | |
| Malik | Dog MI/Pacing | i.v. | 5 | 15 min | ? | +25% | +61% | −17% | +29% | |||
| Bakkehaug | Pig (stunning) | i.v. | 7 | 20 min | 500–1000 | +20% | 0 | 0 | 0 | −17% | −31% | −18% |
| Teerlink | Human Con | i.v. | 34 | 1–24 h | 400–500 | +18% | +11% | −3% | ||||
| Cleland | Human CHF | i.v. | 45 | 1.5–96 h | 400–500 | +19% | +13% | +9% | 0 | |||
| ATOMIC-HF | Human AHF | i.v. | 89 | 48 h | 300–787 | +17%a | 0 | −2 b.p.m. | 0 (EDD) | |||
| COSMIC | Human CHF | oral | 427 | 20 weeks | 318 | +8% | +7% | -4% | 0b | c | −2% (EDD) |
| Study | Species, condition | Appl. | n | Time of OM admin. | OM Plasma Conc. (ng/mL) | SET | SV | HR | C. O. | SVR | LVEDP | LVEDV (LVEDD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shen | Dog MI/Pacing | i.v. | 6 | 15 min–72 h | ? | +20-30% | +44% | −15% | +22% | −15% | −17% | |
| Malik | Dog MI/Pacing | i.v. | 5 | 15 min | ? | +25% | +61% | −17% | +29% | |||
| Bakkehaug | Pig (stunning) | i.v. | 7 | 20 min | 500–1000 | +20% | 0 | 0 | 0 | −17% | −31% | −18% |
| Teerlink | Human Con | i.v. | 34 | 1–24 h | 400–500 | +18% | +11% | −3% | ||||
| Cleland | Human CHF | i.v. | 45 | 1.5–96 h | 400–500 | +19% | +13% | +9% | 0 | |||
| ATOMIC-HF | Human AHF | i.v. | 89 | 48 h | 300–787 | +17%a | 0 | −2 b.p.m. | 0 (EDD) | |||
| COSMIC | Human CHF | oral | 427 | 20 weeks | 318 | +8% | +7% | -4% | 0b | c | −2% (EDD) |
From Shen et al.,84 Malik et al.,83 Bakkehaug et al.,86 Teerlink et al.,16 Cleland et al.,15 ATOMIC-HF,18 and COSMIC-HF.19
C. O., cardiac output; HR, heart rate; ICM, dog model of ischaemic cardiomyopathy; LVEDP, LV end-diastolic pressure; LVEDV(D), LV end-diastolic volume (dimension); n, number of animals/patients; SET, systolic ejection time; SV, stroke volume; SVR, systemic vascular resistance.
Estimated; no baseline SET indicated; assuming a SET of 316 ms as in Cleland et al. (2011).
Estimated from the least square mean changes in SV and HR.
Decrease of NT-proBNP by 970 pg/mL vs. placebo.
Effects of omecamtiv mecarbil on haemodynamics in large animal models and clinical trials
| Study | Species, condition | Appl. | n | Time of OM admin. | OM Plasma Conc. (ng/mL) | SET | SV | HR | C. O. | SVR | LVEDP | LVEDV (LVEDD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shen | Dog MI/Pacing | i.v. | 6 | 15 min–72 h | ? | +20-30% | +44% | −15% | +22% | −15% | −17% | |
| Malik | Dog MI/Pacing | i.v. | 5 | 15 min | ? | +25% | +61% | −17% | +29% | |||
| Bakkehaug | Pig (stunning) | i.v. | 7 | 20 min | 500–1000 | +20% | 0 | 0 | 0 | −17% | −31% | −18% |
| Teerlink | Human Con | i.v. | 34 | 1–24 h | 400–500 | +18% | +11% | −3% | ||||
| Cleland | Human CHF | i.v. | 45 | 1.5–96 h | 400–500 | +19% | +13% | +9% | 0 | |||
| ATOMIC-HF | Human AHF | i.v. | 89 | 48 h | 300–787 | +17%a | 0 | −2 b.p.m. | 0 (EDD) | |||
| COSMIC | Human CHF | oral | 427 | 20 weeks | 318 | +8% | +7% | -4% | 0b | c | −2% (EDD) |
| Study | Species, condition | Appl. | n | Time of OM admin. | OM Plasma Conc. (ng/mL) | SET | SV | HR | C. O. | SVR | LVEDP | LVEDV (LVEDD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shen | Dog MI/Pacing | i.v. | 6 | 15 min–72 h | ? | +20-30% | +44% | −15% | +22% | −15% | −17% | |
| Malik | Dog MI/Pacing | i.v. | 5 | 15 min | ? | +25% | +61% | −17% | +29% | |||
| Bakkehaug | Pig (stunning) | i.v. | 7 | 20 min | 500–1000 | +20% | 0 | 0 | 0 | −17% | −31% | −18% |
| Teerlink | Human Con | i.v. | 34 | 1–24 h | 400–500 | +18% | +11% | −3% | ||||
| Cleland | Human CHF | i.v. | 45 | 1.5–96 h | 400–500 | +19% | +13% | +9% | 0 | |||
| ATOMIC-HF | Human AHF | i.v. | 89 | 48 h | 300–787 | +17%a | 0 | −2 b.p.m. | 0 (EDD) | |||
| COSMIC | Human CHF | oral | 427 | 20 weeks | 318 | +8% | +7% | -4% | 0b | c | −2% (EDD) |
From Shen et al.,84 Malik et al.,83 Bakkehaug et al.,86 Teerlink et al.,16 Cleland et al.,15 ATOMIC-HF,18 and COSMIC-HF.19
C. O., cardiac output; HR, heart rate; ICM, dog model of ischaemic cardiomyopathy; LVEDP, LV end-diastolic pressure; LVEDV(D), LV end-diastolic volume (dimension); n, number of animals/patients; SET, systolic ejection time; SV, stroke volume; SVR, systemic vascular resistance.
Estimated; no baseline SET indicated; assuming a SET of 316 ms as in Cleland et al. (2011).
Estimated from the least square mean changes in SV and HR.
Decrease of NT-proBNP by 970 pg/mL vs. placebo.
In healthy men and patients with stable HF, at comparable increases in SET, however, the net increases in SV, cardiac output and the ensuing decreases in heart rate were overall smaller than in the preclinical studies, perhaps reflecting the broader range of baseline conditions found in human studies (Table 2). In these early studies, OM was studied over a broad range of plasma concentrations, in some cases exceeding 1200 ng/mL. Increases in SET are noted at plasma concentrations as low as 100–200 ng/mL, while the effect on SV appeared to plateau at 400–500 ng/mL. In some individuals, myocardial ischaemia developed with chest pain, ECG changes and/or troponin rises at plasma concentrations beyond 1200 ng/mL.16 , 17 This may be explained by an excessive increase in SET, prolonging cardiac contraction, and progressively shortening diastole (during which coronary perfusion takes place).16 , 17 In a trial of patients with ischaemic cardiomyopathy and angina in daily life, however, OM at target plasma concentrations of 295 ng/mL and 550 ng/mL, respectively, did not affect symptom-limited exercise capacity in treadmill tests or plasma troponin I levels.18 Subsequent trials focused on dose regimens that constrain exposure to less than 1000 ng/mL.
In the ATOMIC-AHF study on patients with AHF and an LVEF ≤40%, the primary endpoint of dyspnoea relief was not reached by three ascending doses of intravenous infusion of OM vs. placebo.19 However, in the highest dose group (n = 202), more patients responded with dyspnoea relief to OM (51%) than to placebo (37%; P = 0.034). In an echocardiographic substudy, OM prolonged SET and decreased LV end-systolic dimension, although LV stroke volume was not increased.19 Additionally, slight decreases in heart rate (−2 b.p.m.) and increases in systolic blood pressure were noted (Table 2).
In the COSMIC-HF trial, oral OM at either a fixed dose (25 mg twice daily) or dosing based on a pharmacokinetic titration protocol was tested against placebo in patients with stable (not acute) systolic HF receiving standard of care therapy.20 After 20 weeks, moderate increases in SET and SV and a slight reduction in heart rate were noted in the pharmacokinetic titration group (Table 2). The latter effect may reflect slightly reduced endogenous sympathetic activity.98 Furthermore, the LV end-diastolic volume decreased by 11 mL and NT-proBNP levels dropped by 970 pg/mL compared to placebo, respectively. As in ATOMIC-AHF,19 there was a small increase in cardiac troponin I that did not correlate with OM plasma concentrations.16 , 17 The frequency of deaths, arrhythmias, hospital admissions, or adverse events was not different between groups, suggesting safety.
Overall, the haemodynamic profile of OM appears promising within its therapeutic range. The increase in cardiac contractility and subsequent prolongation of SET increases LV stroke volume in patients with chronic HF and consequently, blood pressure should rise initially which then may reduce endogenous sympathetic activation. This is indicated by the slight, though consistent lowering of heart rate in human and animal studies. As a result, cardiac output in humans appears largely unchanged despite the modest decrease in heart rate, suggesting improved cardiac efficiency. Furthermore, the decrease in LV filling pressures, as indicated by the decrease in NT-proBNP in COSMIC-HF or the decrease of LV end-diastolic pressures in acute studies in the dog indicate LV unloading that may facilitate reverse remodelling of the LV. The now initiated GALACTIC-HF trial (NCT02929329), which aims to include 8000 patients with chronic HF will eventually clarify the long-term outcome by OM. Meanwhile, some uncertainties remain regarding the mechanisms of action of OM and their implications for cardiac function and long-term outcome.
Diastolic dysfunction
In cardiac myocytes, the increase in systolic function by OM comes at the cost of increased diastolic tension (Figure 6C), indicated by shortening of diastolic cell length. At low (OM) of 200–400 nM, systolic improvement outweighs the diastolic deficit in rat cardiac myocytes, while at 800 nM, this relation reverses. In an in vivo pig model of myocardial stunning after ischaemia/reperfusion, OM reduced both end-diastolic and end-systolic volumes to similar extents (Figure 6E) at concentrations that prolonged SET by 20% (Table 2). Thereby LVEF pseudo-increased, while SV did not.99 Furthermore, the OM-induced increase in cardiac output was smaller in humans with or without HF compared to the dogs with HF (Table 2). In light of the results on post-ischaemic pigs,99 it needs to be considered whether an improvement of SET (and therefore, SV) by OM may have been (partly) offset by decreases in end-diastolic volume or filling. The improvement of NT-proBNP by long-term OM in COSMIC HF 20 however, rather argues against a meaningful deterioration of diastolic function by these doses of OM.
Bioenergetic aspects
In dogs with HF, OM did not increase O2 consumption97 although in the post-ischaemic pig model, O2 consumption tended to increase.99 In isolated mouse hearts, OM impaired myocardial efficacy by increasing O2 consumption in working hearts and during basal (resting) metabolism, which was abolished by a myosin-ATPase inhibitor.99 These data suggest that OM increases (tonic) myosin-ATPase activity100 and thereby O2 consumption, which however contrasts with the effect of OM to inhibit the basal ATPase of myosin in vitro.101 In skinned rat cardiac myocytes, OM shifted the pCa/force relationship to the left, indicative of sensitizing myofilaments to Ca2+ (Figure 6F).102 In human myocardium, OM increased the myosin duty ratio which resulted in enhanced Ca2+ sensitivity, but slower force development.103 In a mouse model of dilated cardiomyopathy with decreased myofilament Ca2+ sensitivity, OM resensitized myofilaments towards control levels.104 However, in the majority of patients with HF, the Ca2+ affinity of the myofilaments is increased rather than decreased,28–30 , 105 , 106 and in LV myocardium of patients with terminal HF, increased diastolic tension consumes as much ATP and O2 as systolic tension, and elevated diastolic tension is a substantial energetic burden in failing hearts especially at higher heart rates.107
In conclusion, whether OM has a neutral or even net energy-sparing effect on myocardial bioenergetics, or whether the drug’s net effect on myosin could increase O2-consumption is a question future research should continue to address. Furthermore, the development of small molecules targeting sarcomeric motor proteins is an emerging field that is discussed in more detail in the Supplementary material online.
Alternative treatments targeting excitation–contraction coupling
Nitroxyl
Nitroxyl (HNO) is produced by NO synthase under conditions of oxidative or nitrosative stress. HNO donated by Angeli’s salt (AS) improves cardiac function in normal and failing dogs, independently of β-AR signalling, with no change in cGMP levels.108 , 109 In cardiac myocytes, HNO increases fractional shortening (FS) and Ca2+ transients with no involvement of cAMP/PKA or cGMP/protein kinase G signalling.110 Instead, HNO modifies cysteine residues to enhance Ca2+ handling and increase myofilament Ca2+ sensitivity. In particular, HNO alters the inhibitory interaction between phospholamban and SERCA2a in a redox-dependent manner, improving SR Ca2+ uptake and release in isolated myocytes/hearts (Figure 7).111 , 112 In addition, HNO modifies the actin–tropomyosin and myosin heavy chain-myosin light chain 1 interactions, increasing Ca2+ sensitivity and force generation in intact and skinned muscles (Figure 7).113
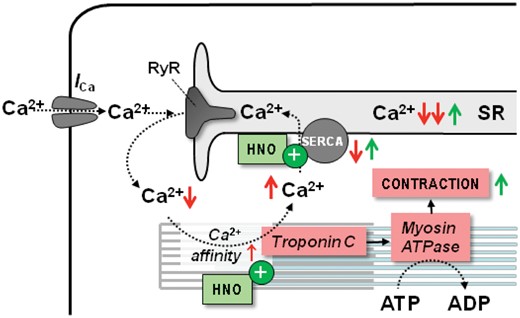
Mechanisms of action of nitroxyl (HNO) in HF. HNO affects redox-sensitive residues of various proteins involved in myocyte Ca2+ handling. In particular, HNO increases SERCA activity and sensitizes myofilaments to Ca2+. In concert, these properties increase SR Ca2+ load, systolic Ca2+ transients and contraction. Red arrows ( ↑↓) indicate the direction of change in heart failure, while green arrows ( ↑↓) indicate the direction induced by HNO.
Whereas beneficial effects of NO signalling to cGMP may be lost in conditions of cardiac oxidative stress (by the high reactivity of NO with ROS), this does not occur with HNO, whose efficacy is preserved in conditions with altered redox balance.114 , 115 Novel HNO donors (e.g. CXL-1020) are long-lasting and more specific. CXL-1020 has HNO-dependent positive inotropic and lusitropic effects in isolated cardiac myocytes in vitro and in whole animal studies in vivo, in both normal and failing conditions. Human Phase I–IIa clinical trials were recently completed (NCT01092325, NCT01096043). In patients with systolic HF, CXL-1020 reduced both left and right heart filling pressures and SVR, while increasing SV. Heart rate was unchanged, and arterial pressure declined modestly.116 Currently, an improved second-generation HNO donor, CXL-1427, is tested in Phase I and II trials on healthy volunteers and patients with HF (NCT02157506, NCT02819271).
Other compounds and interventions
The mechanisms and, where appropriate, clinical results of Istaroxime, SERCA2a gene therapy and EF-hand proteins are discussed in the Supplementary material online.
Energetic considerations
Taken together, inotropic agents have three principal modes of action:
Activation of the adrenergic system,
sensitization of myofilaments to Ca2+ and
reconstitution of cytosolic Ca2+ handling independent of adrenergic activation.
The modes of action of inotropic drugs have important energetic consequences. Most cellular ATP is consumed by SERCA, NKA, and myosin ATPase.117 A central mechanism to match ATP supply to demand is ‘parallel activation’ by Ca2+,31 where Ca2+ activates both ATP-consumption and -regeneration (Figures 3 and 8): Increases in [Ca2+]c accelerate ATP consumption by EC coupling, hastening respiration via ADP which oxidizes NADH and FADH2 at the respiratory chain. On the other hand, Ca2+ enters mitochondria to activate Krebs cycle dehydrogenases, accelerating NADH and FADH2 regeneration (Figure 8).31 Consequently, hormones or drugs that increase Ca2+ handling (e.g. catecholamines) in the short-term induce this ‘parallel activation’ of respiration, maintaining the redox state of NADH and FADH2 in normal hearts (Figure 8).31 In the failing heart, however, mitochondrial Ca2+ uptake is impaired, resulting in NADH oxidation during β-adrenergic stimulation.31 , 33 Since NADH is coupled to the NADPH pool, and NADPH required for ROS detoxification (Figures 3 and 8), a mismatch between cardiac work and mitochondrial Ca2+ uptake induces oxidative stress.31 , 33 This may contribute to arrhythmias, systolic dysfunction, and maladaptive remodelling through necrosis and other redox-sensitive signalling pathways.33 Therefore, improving SR Ca2+ content and release, as has been observed with AAV1/SERCA2a118 or HNO110 in animal models of HF, may improve the efficiency of mitochondrial Ca2+ uptake and make the failing heart less sensitive towards β-AR-mediated oxidative stress and damage. In patients with improved LVEF in response to β-blockers, SERCA gene expression was strongly upregulated,119 suggesting that also β-blockers may improve mitochondrial redox regulation through restoring defective EC coupling in the long term.
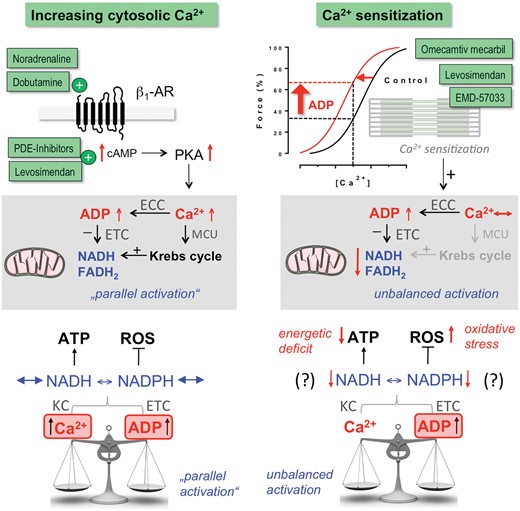
Known and hypothesized bioenergetic consequences of inotropic interventions that either increase cytosolic Ca2+ or myofilament Ca2+ sensitivity. ECC, excitation–contraction coupling; ETC, electron transport chain; MCU, mitochondrial Ca2+ uptake.
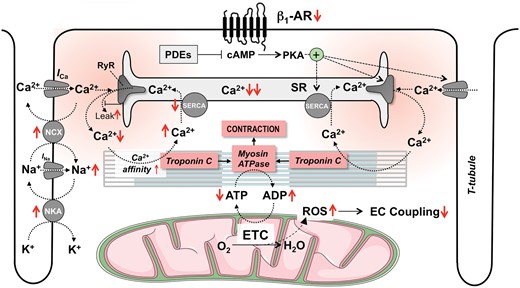
Mechanisms of excitation-contraction coupling, known defects in heart failure and which targets inotropic compounds have. In systolic HF, contractile dysfunction is primarily related to attenuated increases in cytosolic Ca2+ during systole. This is the result of decreased activity of the sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA) and leaky ryanodine receptors (RyR). Maximal contractility is further limited by decreased adenosine triphosphate (ATP) production in mitochondria. Dobutamine and norepinephrine activate β1-adrenergic receptors (β1-AR), increasing cAMP which phosphorylates protein kinase A (PKA). PKA in turn phosphorylates proteins involved in EC coupling and thereby accelerates the kinetics and amounts of cytosolic Ca2+ during systole. The phosphodiesterase 3 (PDE3) inhibitors enoximone and milrinone potentiate β-AR-induced cAMP elevations and therefore, have similar effects on inotropy as dobutamine, increasing Ca2+ fluxes. Digitalis inhibits the Na+/K+-ATPase (NKA) and thereby elevates intracellular Na+, which in turn elevates Ca2+ by hampering cytosolic Ca2+ export via the forward mode Na+/Ca2+ exchanger (NCX) and may increase reverse mode NCX-mediated Ca2+ influx during the early phase of the action potential. Istaroxime has similar effects as digitalis but also activates SERCA to accelerate diastolic Ca2+ uptake into the SR. SERCA2a gene therapy increases the mRNA and protein levels of SERCA and thereby, potentiates SR Ca2+ uptake and release. Nitroxyl (HNO) activates SERCA activity and increases myofilament Ca2+ sensitivity. Levosimendan increases the affinity of troponin C to Ca2+ and thereby, increases force generation for any given cytosolic Ca2+ concentration. In addition, levosimendan inhibits PDE3, which elevates cAMP and PKA activity with subsequent effects on Ca2+ handling as described above for catecholamines and PDE-inhibitors. Omecamtiv mecarbil is a myosin activator that prolongs actin–myosin interaction and thereby, results in a prolongation (but not acceleration) of contraction. Elamipretide (also known as Bendavia or MTP-131) binds to cardiolipin in the inner mitochondrial membrane, increasing ATP production and reducing the generation of reactive oxygen species (ROS). Trimetazidine and perhexiline optimize substrate utilization and thereby, improve cardiac energetics. Iron restores iron deficiency and thereby, may improve function of Krebs cycle enzymes and possibly, the electron transport chain (ETC).
Conversely, when sensitizing myofilaments to Ca2+, the increase in ATP consumption at the myofilaments may not be adequately matched by Ca2+-induced Krebs cycle activation in mitochondria (Figure 8). In fact, pre-stretching cardiac trabeculae increases force production independent of an increase in Ca2+ via the Frank–Starling mechanism, which is related to myofilament Ca2+ sensitization, and this oxidizes NADH.120 Whether such energetic mismatch and possibly further downstream consequences such as energetic deficit and oxidative stress are also the result of Ca2+ sensitization through inotropic drugs (i.e. EMD-57033, levosimendan, OM, nitroxyl etc.), mutations occurring in patients with HCM75 , 76 and/or post-translational modifications occurring in patients with systolic HF106 needs to be clarified by future research. In the context of inotropic drugs, however, a profile where Ca2+ sensitization is coupled to restoration of Ca2+ handling (i.e. nitroxyl) or increases of Ca2+ (i.e. levosimendan; Table 3) may ameliorate energetic/redox mismatch occurring through Ca2+ sensitization per se.
Mechanisms of action of clinically tested agents targeting inotropy
| Drug/mechanism | Increasing cAMP | Myofilament Ca2+ sensitization | Restoring cytosolic Ca2+ handling |
|---|---|---|---|
| Dobutamine | +++ | ||
| Milrinone | +++ | ||
| Levosimendan | ++ | ++ | |
| EMD-57033 | (+) | +++ | |
| Omecamtiv mecarbil | +++ (a) | ||
| Nitroxyl (HNO) | + | ++ | |
| AAV1/SERCA2a | +++ | ||
| EF-hand Ca2+-binding motifs | +++ |
| Drug/mechanism | Increasing cAMP | Myofilament Ca2+ sensitization | Restoring cytosolic Ca2+ handling |
|---|---|---|---|
| Dobutamine | +++ | ||
| Milrinone | +++ | ||
| Levosimendan | ++ | ++ | |
| EMD-57033 | (+) | +++ | |
| Omecamtiv mecarbil | +++ (a) | ||
| Nitroxyl (HNO) | + | ++ | |
| AAV1/SERCA2a | +++ | ||
| EF-hand Ca2+-binding motifs | +++ |
Principle mechanism is myosin activation, but this increases myofilament Ca2+ sensitivity as well.89–91
Mechanisms of action of clinically tested agents targeting inotropy
| Drug/mechanism | Increasing cAMP | Myofilament Ca2+ sensitization | Restoring cytosolic Ca2+ handling |
|---|---|---|---|
| Dobutamine | +++ | ||
| Milrinone | +++ | ||
| Levosimendan | ++ | ++ | |
| EMD-57033 | (+) | +++ | |
| Omecamtiv mecarbil | +++ (a) | ||
| Nitroxyl (HNO) | + | ++ | |
| AAV1/SERCA2a | +++ | ||
| EF-hand Ca2+-binding motifs | +++ |
| Drug/mechanism | Increasing cAMP | Myofilament Ca2+ sensitization | Restoring cytosolic Ca2+ handling |
|---|---|---|---|
| Dobutamine | +++ | ||
| Milrinone | +++ | ||
| Levosimendan | ++ | ++ | |
| EMD-57033 | (+) | +++ | |
| Omecamtiv mecarbil | +++ (a) | ||
| Nitroxyl (HNO) | + | ++ | |
| AAV1/SERCA2a | +++ | ||
| EF-hand Ca2+-binding motifs | +++ |
Principle mechanism is myosin activation, but this increases myofilament Ca2+ sensitivity as well.89–91
Finally, it should be considered whether targeting mitochondria may be an alternative indirect inotropic intervention, since in dogs with HF, elamipretide—which accumulates in mitochondria and improves mitochondrial function—acutely increased cardiac output by a similar extent (+25%)121 as OM (+22 and +29%, respectively; Table 2).96 , 97 Furthermore, trimetazidine and perhexiline target substrate metabolism of mitochondria and improve LVEF, haemodynamics, cardiac energetics, and symptoms in patients with HF (for more details on these compounds see Supplementary material online).122–126 Finally, cardiac myocyte-specific ID impaired the response to dobutamine in preclinical models of HF which could be restored by iron supplementation,127 suggesting that pharmacological restoration of mitochondrial function may also regenerate the heart’s response to inotropic stimulation.
Summary
Catecholamines and PDE-inhibitors are associated with excess mortality presumably related to the induction of arrhythmias in the short-term and the activation of signalling pathways that aggravate maladaptive remodelling of the failing heart in the long-term. Although levosimendan has so far been viewed as a Ca2+ sensitizer, its inotropic effect relies on PDE3-inhibition as well. As a myosin activator, OM improves systolic function without activating adrenergic signalling or increasing cytosolic Ca2+, but its therapeutic range is limited by diastolic dysfunction at higher doses. Nitroxyl restores cytosolic Ca2+ handling in failing hearts without activating cAMP-dependent signalling pathways and shows a promising haemodynamic profile, but its clinical usefulness awaits further clinical testing. Besides the impact of adrenergic signalling, bioenergetic aspects need to be considered to estimate the comprehensive profile and long-term consequences of any agent that affects inotropy.
Statements and recommendations
Based on these preclinical and clinical data as well as the bioenergetic considerations, the Committees on Translational Research and on Acute Heart Failure of the HFA of the ESC make the following statements and recommendations:
Currently available drugs primarily targeting inotropy are cardiotropic glycosides, catecholamines (in particular, dobutamine), PDE-inhibitors, and levosimendan.
According to the current HF Guidelines,3 the use of catecholamines, PDE-inhibitors and levosimendan should be limited to patients with hypotension (SBP <90 mmHg) and/or signs/symptoms of hypoperfusion despite adequate filling status to increase cardiac output and improve peripheral perfusion to maintain end-organ function (IIb, C).
PDE-inhibitors and levosimendan can cause hypotension due to vasodilatory actions.
Vasopressors (preferably norepinephrine) may be considered to increase blood pressure and vital organ perfusion in patients with cardiogenic shock despite the use of inotropes (IIb, B).3
So far, all inotropic drugs recommended for the use in patients with acute HF (including levosimendan) activate adrenergic signalling at least to some extent.
Longer-term use of drugs that exclusively target adrenergic signalling (catecholamines, PDE-inhibitors) are associated with adverse outcome.
Levosimendan, with its hybrid Ca2+ sensitization and adrenergic action (PDE3 inhibition), could be useful in selected patient populations, which may include patients treated with β-blockers and patients with advanced HF in an outpatient setting (repeated dosing) to reduce hospitalization and improve quality of life, although this requires additional proof from larger trials.
Since mitochondrial function is intimately linked to cellular Ca2+ handling,31 the bioenergetic consequences of treatments targeting inotropy need to be considered to understand their short- and long-term consequences.
On theoretical grounds, treatments that restore the defects of cytosolic Ca2+ handling in the failing heart without activating adrenergic signalling may be a promising avenue since they avoid diastolic dysfunction and potentially bioenergetic mismatch of pure Ca2+ sensitization, but also adverse long-term consequences of adrenergic activation.
Future research should be directed towards deepening our understanding of the close interplay between EC coupling and mitochondrial energetics, since only the integration of these aspects will resolve the net biological effects of drugs targeting inotropy in the short- and long-term.
Acknowledgements
We thank Gerasimos Filippatos, Fady I. Malik, and Piero Pollesello for insightful discussions and valuable input to the manuscript.
Funding
C.M. is supported by the Deutsche Forschungsgemeinschaft (DFG; SFB 894, TRR-219, and Ma 2528/7-1), the German Federal Ministry of Education and Science (BMBF; 01EO1504) and the Corona foundation. J.M.M. is supported by grants from the NIH. C.G.T. is supported by grants of Federico II University-Ricerca d Ateneo. J.L.B. is supported by Fonds National de la Recherche Scientifique and European Union (UE Horizon2020 GA634559. A.D. is supported by the German Cardiac Society (DGK) and institutional research grants of the University Hospital Regensburg (ReForM-A/B). C.Mu. received research grants from the Swiss National Science Foundation, the Swiss Heart Foundation, the European Union, the Cardiovascular Research Foundation Basel, Basel University and the University Hospital Basel. W.H.Z. is supported by the DZHK (German Center for Cardiovascular Research), the BMBF, the DFG (ZI 708/10-1, SFB 937 A18, SFB 1002 C04/01 and IRTG 1816 RP12), and Foundation Leducq.
Conflict of interest: C.M. serves as an advisor to Servier and received speaker honoraria from Servier, Boehringer Ingelheim, Bayer, Bristol Myers Squibb, Pfizer, Daiichi Sankyo, Novartis and Berlin Chemie. T.E. is co-founder of spin-off company EHT Technologies GmbH. F.R.H. received grants from Sanofi, Novartis and Menarini. A.R.L. reports personal fees from Novartis, AMGEN and Servier. D.J.M. holds Patent EP2277124 B1 issued to Medizinische Hochschule Hannover and TU Dresden. J.M.M. has a patent pending. Z.P. received speaker honoraria from Orion Pharma. C.G.T. received speaker honoraria from Alere and is co-inventor of the Canadian patent no. 2,613,477: “Thiol Sensitive Positive Inotropes”, issued on Dec 3, 2013. M.B.Y. reports on institutional honoraria from Novartis, Bayer Healthcare and Amgen. S.D.A. received fees for trial / registry steering committee work and advisory boards from Bayer, Boehringer Ingelheim, Novartis, Servier, Stealth, and Vifor, and reports on grants for IITs from Abbott Vascular and Vifor. J.L.B. is advisor to Sanofi and Amgen. J.B. received personal fees from Orion, Novartis and Abiomed. J.G.C. serves in the steering committee for GALACTIC (Amgen/Cytokinetics) and is Chief Investigator for IDDEA-HF (Stealth Biopharmaceuticals). He received grants and/or personal fees from Novartis and Servier. R.A.d.B. is a minority shareholder of scPharmaceuticals, Inc., and received personal fees from MandalMed Inc, Novartis, and Servier. The UMCG, which employs R.A.d.B., has received research grants and/or fees from AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Roche, Trevena, and ThermoFisher GmbH. V.P.H. received consultation fees from Orion Pharma. L.H.L. received research grants and/or consultation fees from Orion Pharma, Amgen, Novartis, Boehringer Ingelheim, Vifor Pharma, Astra Zeneca, Merck and Sanofi. J.M. is consultant for Cardiorentis, advisor for Novartis, and received travel grants from Boehringer Ingelheim, Novartis and Menarini. M.M. received consulting honoraria from Amgen, Bayer, Novartis and Servier. C.Mu. received research grants from Abbott, ALERE, Astra Zeneca, Beckman Coulter, Biomerieux, BRAHMS, Critical Diagnostics, Ortho Diagnostics, Roche, Siemens and Singulex, as well as speaker/consulting honoraria from Abbott, ALERE, Astra Zeneca, Biomerieux, BMS, Boehringer Ingelheim, BRAHMS, Cardiorentis, Duke University, Novartis, Roche, Sanofi, Singulex, Siemens, and Zurich Heart House. A.R. received grants from Servier, Actavis, Boehringer Ingelheim, Astra Zeneca and Bayer, and reports on personal fees from Pfizer, Merck, Berlin Chemie, AstraZeneca, Hemofarm Stada, Krka Pharma and Roche Diagnostics. F.R. reports on personal fees (talks) from SJM, Novartis, Servier, Zoll, Bayer and Abbott and serves as an advisor to AstraZeneca, Sanofi, Amgen, Roche, Pfizer and BMS. He received honoraria of steering committee meetings from Fresenius, Vifor and Cardiorentis. W.H.Z. received honoraria for lectures from Daiichi-Sankyo and is founder and advisor of myriamed GmbH and Repairon GmbH. A.M. received honoraria (lectures) from Orion, Servier, Abbott, Novartis, and is a consultant for BMS, Cardiorentis, Roche and Sphyngotec. All other authors declared no conflict of interest.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}