





Abstract
The benefit of the β1-adrenergic receptor (β1-AR) agonist dobutamine for treatment of acute heart failure in peripartum cardiomyopathy (PPCM) is controversial. Cardiac STAT3 expression is reduced in PPCM patients. Mice carrying a cardiomyocyte-restricted deletion of STAT3 (CKO) develop PPCM. We hypothesized that STAT3-dependent signalling networks may influence the response to β-AR agonist treatment in PPCM patients and analysed this hypothesis in CKO mice.
Follow-up analyses in 27 patients with severe PPCM (left ventricular ejection fraction ≤25%) revealed that 19 of 20 patients not obtaining dobutamine improved cardiac function. All seven patients obtaining dobutamine received heart transplantation (n = 4) or left ventricular assist devices (n = 3). They displayed diminished myocardial triglyceride, pyruvate, and lactate content compared with non-failing controls. The β-AR agonist isoproterenol (Iso) induced heart failure with high mortality in postpartum female, in non-pregnant female and in male CKO, but not in wild-type mice. Iso induced heart failure and high mortality in CKO mice by impairing fatty acid and glucose uptake, thereby generating a metabolic deficit. The latter was governed by disturbed STAT3-dependent signalling networks, microRNA-199a-5p, microRNA-7a-5p, insulin/glucose transporter-4, and neuregulin/ErbB signalling. The resulting cardiac energy depletion and oxidative stress promoted dysfunction and cardiomyocyte loss inducing irreversible heart failure, which could be attenuated by the β1-AR blocker metoprolol or glucose-uptake-promoting drugs perhexiline and etomoxir.
Iso impairs glucose uptake, induces energy depletion, oxidative stress, dysfunction, and death in STAT3-deficient cardiomyocytes mainly via β1-AR stimulation. These cellular alterations may underlie the dobutamine-induced irreversible heart failure progression in PPCM patients who frequently display reduced cardiac STAT3 expression.
• The benefit of the β1-adrenergic receptor (β1-AR) agonist dobutamine for treatment of acute heart failure in peripartum cardiomyopathy (PPCM) is controversial. Cardiac STAT3 expression is reduced in PPCM patients, and mice with a cardiomyocyte-restricted deletion of STAT3 develop PPCM.
• Data from the German PPCM registry show that dobutamine treatment in patients with severe PPCM was associated with an adverse outcome (heart transplantation or left ventricular assist device implantation) while almost all patients not receiving dobutamine recovered.
• β1-AR stimulation with isoproterenol in mice lacking cardiomyocyte STAT3 induced severe cardiac dysfunction and high mortality. In STAT3-deficient cardiomyocytes β1-AR stimulation impaired glucose uptake and subsequently induced energy depletion, oxidative stress, dysfunction, and death.
• Our study provides novel insights how dobutamine in the pathophysiology of PPCM may induce irreversible heart failure progression and suggests how the glucose-uptake-enhancing drug perhexiline could help to prevent this.
Introduction
Peripartum cardiomyopathy (PPCM) is a non-familial cardiomyopathy presenting with heart failure (HF) secondary to left ventricular (LV) systolic dysfunction towards the end of pregnancy or in the months following delivery, when no other cause of HF is found.1,2 Onset of the disease is often sudden and in severe cases associated with cardiogenic shock. However, partial or full recovery is frequently possible.1,2 The benefit of dobutamine treatment in PPCM patients with acute HF is controversial and may be associated with adverse outcome.2 Here, we evaluated clinical data sets from the German PPCM registry and observed that all PPCM patients treated with dobutamine required either cardiac transplantation or LV assist devices (LVAD), while in 95% of patients not receiving dobutamine despite similar initial impairment of LV function, cardiac function recovered without the need of cardiac support or transplantation.
Reduced cardiac STAT3 expression was observed in PPCM patients and may play a causal role in disease pathogenesis since mice with a cardiomyocyte-specific deletion of Stat3 (aMHC-Cretg/+;Stat3fl/fl; CKO) develop PPCM.3 During the peripartum phase, STAT3 protects the maternal heart from excessive oxidative stress and the subsequent generation of a disease-promoting angiostatic fragment of the nursing hormone prolactin (16 kDa-PRL), which compromises the cardiac microvasculature.3,4 Here, we hypothesized that STAT3 may also protect the maternal heart from adverse effects caused by β1-adrenergic receptor (β1-AR) activation.
Indeed, the β-AR agonist isoproterenol (Iso) caused HF and high mortality in postpartum as well as nulli-pari CKO mice mainly by inducing metabolic deficits through impairing fatty acid and glucose uptake. The latter was governed by a disturbed STAT3-dependent signalling network involving microRNA-199a-5p (miR-199a) and microRNA-7a-5p (miR-7a), insulin/glucose transporter-4 (GLUT4), and neuregulin (NRG)/ErbB signalling. The resulting cardiac energy depletion impairs the regeneration of mitochondrial pyridine nucleotides, which decreased ATP production and increased emission of reactive oxygen species (ROS) causing functional impairment and cardiomyocyte necrosis and with this irreversible HF. This could be attenuated by the β1-AR blocker metoprolol or the glucose-uptake-promoting drug perhexiline5 that is currently tested in clinical studies.6
Therefore, diminished cardiac STAT3 expression may render PPCM patients more vulnerable to β1-AR agonists-induced cardiac damage and HF progression.
Methods
Expanded methods are available in the Supplementary material online.
Results
Dobutamine treatment is associated with an adverse outcome in peripartum cardiomyopathy patients with severe heart failure
Twenty-seven PPCM patients were selected from the German PPCM registry7 according to a baseline LV ejection fraction (LVEF) ≤25% and the presence of clinical follow-up data (9 ± 4 months). During acute HF, 7 of these patients were treated with dobutamine (Dob-group) while 20 did not obtain dobutamine (no-Dob-group). The decision to use dobutamine was left to the discretion of the treating physicians. The clinical presentation at diagnosis was similar between both groups, while age, gravida, and parity were higher in the no-Dob-group compared with the Dob-group (Table 1). Patients in the no-Dob-group were more frequently treated with a β-blocker (n = 19 out of 20) compared with the Dob-group (n = 4 out of 7, P =0.042), while the proportion of patients receiving an angiotensin-converting enzyme inhibitor/angiotensin receptor blocker or bromocriptine was similar (Table 1). All seven patients in the Dob-group experienced an adverse outcome, i.e. LVAD implantation (n = 3) or heart transplantation (n = 4) and/or subsequent death (n = 1). In contrast, 95% of the no-Dob-group improved LVEF (baseline, 18 ± 5%; follow-up, 46 ± 11%; P <0.0001), while only one patient (5%) underwent heart transplantation (P <0.0001 vs. Dob-group; two-sided Fisher's exact test).
Clinical parameters, echocardiographic findings, and laboratory tests in peripartum cardiomyopathy patients at diagnosis
| Parameters | No dobutamine (n = 20) | Dobutamine (n = 7) | P-value (no dobutamine vs. dobutamine) |
|---|---|---|---|
| Age (years) (mean ± SD) | 36 ± 4 | 28 ± 4 | 0.005 |
| Gravida median (range) | 3 (1–4) | 1.5 (1–2) | 0.008 |
| Parity median (range) | 2 (1–3) | 1 (1–2) | 0.011 |
| LVEF (%) (mean ± SD) | 18 ± 5 | 20 ± 5 | 0.493 |
| LVEDD (mm) (mean ± SD) | 64 ± 6 | 70 ± 12 | 0.306 |
| Heart rate (b.p.m.) (mean ± SD) | 88 ± 18 | 111 ± 21 | 0.079 |
| NT-proBNP (pg/mL) median (range) | 4225 (1158–11484) | 4436 (2518–12390) | 0.868 |
| β-Blocker | 19 (90%) | 4 (57%) | 0.042 |
| ACE-inhibitor/ARB | 20 (100%) | 6 (86%) | 0.259 |
| Bromocriptine | 18 (86%) | 4 (57%) | 0.091 |
| Parameters | No dobutamine (n = 20) | Dobutamine (n = 7) | P-value (no dobutamine vs. dobutamine) |
|---|---|---|---|
| Age (years) (mean ± SD) | 36 ± 4 | 28 ± 4 | 0.005 |
| Gravida median (range) | 3 (1–4) | 1.5 (1–2) | 0.008 |
| Parity median (range) | 2 (1–3) | 1 (1–2) | 0.011 |
| LVEF (%) (mean ± SD) | 18 ± 5 | 20 ± 5 | 0.493 |
| LVEDD (mm) (mean ± SD) | 64 ± 6 | 70 ± 12 | 0.306 |
| Heart rate (b.p.m.) (mean ± SD) | 88 ± 18 | 111 ± 21 | 0.079 |
| NT-proBNP (pg/mL) median (range) | 4225 (1158–11484) | 4436 (2518–12390) | 0.868 |
| β-Blocker | 19 (90%) | 4 (57%) | 0.042 |
| ACE-inhibitor/ARB | 20 (100%) | 6 (86%) | 0.259 |
| Bromocriptine | 18 (86%) | 4 (57%) | 0.091 |
LVEF and LV end-diastolic diameter (LVEDD, echocardiography), heart rate (HR, b.p.m., ECG), and NT-proBNP were determined at diagnosis. Comparison of means between groups was performed by unpaired t-test after testing for normal distribution using the Kolmogorov–Smirnov test. The proportion of patients within each group receiving a particular medication (β-blocker, ACE-inhibitor/ARB, or bromocriptine) was performed by Fisher's exact test.
Clinical parameters, echocardiographic findings, and laboratory tests in peripartum cardiomyopathy patients at diagnosis
| Parameters | No dobutamine (n = 20) | Dobutamine (n = 7) | P-value (no dobutamine vs. dobutamine) |
|---|---|---|---|
| Age (years) (mean ± SD) | 36 ± 4 | 28 ± 4 | 0.005 |
| Gravida median (range) | 3 (1–4) | 1.5 (1–2) | 0.008 |
| Parity median (range) | 2 (1–3) | 1 (1–2) | 0.011 |
| LVEF (%) (mean ± SD) | 18 ± 5 | 20 ± 5 | 0.493 |
| LVEDD (mm) (mean ± SD) | 64 ± 6 | 70 ± 12 | 0.306 |
| Heart rate (b.p.m.) (mean ± SD) | 88 ± 18 | 111 ± 21 | 0.079 |
| NT-proBNP (pg/mL) median (range) | 4225 (1158–11484) | 4436 (2518–12390) | 0.868 |
| β-Blocker | 19 (90%) | 4 (57%) | 0.042 |
| ACE-inhibitor/ARB | 20 (100%) | 6 (86%) | 0.259 |
| Bromocriptine | 18 (86%) | 4 (57%) | 0.091 |
| Parameters | No dobutamine (n = 20) | Dobutamine (n = 7) | P-value (no dobutamine vs. dobutamine) |
|---|---|---|---|
| Age (years) (mean ± SD) | 36 ± 4 | 28 ± 4 | 0.005 |
| Gravida median (range) | 3 (1–4) | 1.5 (1–2) | 0.008 |
| Parity median (range) | 2 (1–3) | 1 (1–2) | 0.011 |
| LVEF (%) (mean ± SD) | 18 ± 5 | 20 ± 5 | 0.493 |
| LVEDD (mm) (mean ± SD) | 64 ± 6 | 70 ± 12 | 0.306 |
| Heart rate (b.p.m.) (mean ± SD) | 88 ± 18 | 111 ± 21 | 0.079 |
| NT-proBNP (pg/mL) median (range) | 4225 (1158–11484) | 4436 (2518–12390) | 0.868 |
| β-Blocker | 19 (90%) | 4 (57%) | 0.042 |
| ACE-inhibitor/ARB | 20 (100%) | 6 (86%) | 0.259 |
| Bromocriptine | 18 (86%) | 4 (57%) | 0.091 |
LVEF and LV end-diastolic diameter (LVEDD, echocardiography), heart rate (HR, b.p.m., ECG), and NT-proBNP were determined at diagnosis. Comparison of means between groups was performed by unpaired t-test after testing for normal distribution using the Kolmogorov–Smirnov test. The proportion of patients within each group receiving a particular medication (β-blocker, ACE-inhibitor/ARB, or bromocriptine) was performed by Fisher's exact test.
Chronic β1-adrenergic receptor stimulation induces heart failure in postpartum mice with cardiomyocytes-specific STAT3 deletion
![Detrimental effects of Iso stimulation in CKO mice. (A) LV-systolic function (%FS) and (B) representative echocardiographic pictures in end-systolic (upper panel) and end-diastolic (lower panel) parasternal long axis of wild-type and CKO females treated with NaCl, Iso, or Iso and bromocriptin postpartum for 14 days (n = 6–9). (C) Kaplan–Meier survival curve during Iso stimulation [10–30 mg/kg/day; n = 5–29, log-rank (Mantel–Cox) test]. (D) Western blots for p-troponin I and troponin I and (E) corresponding quantification of p-troponin I to troponin I ratio (n = 6–9). (F) Sirius red staining and (G) corresponding fibrosis quantification (n = 5–7, wild-type baseline set as 1), (H) cardiomyocyte necrosis (Evans Blue: necrotic cells, red; wheat germ agglutinin: membranes, green; 4',6-diamidino-2-phenylindole staining: nuclei, blue), and (I) necrosis quantification (n = 3–7), (J) inflammation using the pan-inflammatory marker CD45 (brown; counterstaining with eosin, pink) and (K) quantification of inflammation (n = 4–8, wild-type baseline set as 1). Data are mean ± SD. Scale bars = 100 µm. P-values were computed by two-way ANOVA followed by Bonferroni post hoc test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/38/5/10.1093_eurheartj_ehw086/2/m_ehw08601.jpeg?Expires=1747893775&Signature=ZFTS4EwZXQytQxdgOdGBKHFTw9yZt4Qejw-oHTAqJ30oytqgJlhqCqWAIqPgF5bZEbvEZ6rf5aGyKnQrBex18CqLHgJWLPWI8xFH2COB4xLnd-Nj7bmqTevq4PSlHCCKzCijTmZ-8jNRpKwjKYh5DwWhPrdXGO26htzmyUM5AUvl1J46JXu1fKRjx72y4BKgugiYwRqlC-Xshd~6kFUDYLYkEq6Wk39QD0kZndWBL4fCYQwfHB~DMhdY5sC-lJDMwMBR~oZ5LTu6O7L8J6rGfOkT3iE-F0thuvj4QSMkp6BnelEa9MJZcutix5dsFXHONhZSe8S7gXquZa~VGWoL0g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Detrimental effects of Iso stimulation in CKO mice. (A) LV-systolic function (%FS) and (B) representative echocardiographic pictures in end-systolic (upper panel) and end-diastolic (lower panel) parasternal long axis of wild-type and CKO females treated with NaCl, Iso, or Iso and bromocriptin postpartum for 14 days (n = 6–9). (C) Kaplan–Meier survival curve during Iso stimulation [10–30 mg/kg/day; n = 5–29, log-rank (Mantel–Cox) test]. (D) Western blots for p-troponin I and troponin I and (E) corresponding quantification of p-troponin I to troponin I ratio (n = 6–9). (F) Sirius red staining and (G) corresponding fibrosis quantification (n = 5–7, wild-type baseline set as 1), (H) cardiomyocyte necrosis (Evans Blue: necrotic cells, red; wheat germ agglutinin: membranes, green; 4',6-diamidino-2-phenylindole staining: nuclei, blue), and (I) necrosis quantification (n = 3–7), (J) inflammation using the pan-inflammatory marker CD45 (brown; counterstaining with eosin, pink) and (K) quantification of inflammation (n = 4–8, wild-type baseline set as 1). Data are mean ± SD. Scale bars = 100 µm. P-values were computed by two-way ANOVA followed by Bonferroni post hoc test.
Chronic Iso stimulation induces heart failure in non-pregnant STAT3-deficient mice mainly by β1-adrenergic receptor activation
To investigate whether STAT3 may be important for cardioprotection during β-AR treatment independent of pregnancy and pregnancy hormones, male CKO mice were subjected to Iso treatment (10–30 mg/kg/day) for 2 weeks, which led to a dose-dependent increase in mortality compared with WT siblings (Figure 1C). In CKO mice, Iso treatment induced LV dysfunction and increased LV dilatation with increased heart/body weight ratios (Table 2; Supplementary material online, Figure S1) and cardiomyocyte lengths (CML: +33 ± 14%, *P =0.0001 vs. CKO NaCl; *P indicates two-way ANOVA) with no significant increase in cardiomyocyte cross-sectional area (CSA: +32 ± 28%, *P =0.0861 vs. CKO NaCl). In WT and heterozygous (aMHC-Cretg/+; Stat3fl/+) mice LV function remained compensated (Table 2; Supplementary material online, Figures S1 and S2). Iso-treated WT mice displayed ventricular hypertrophy (Supplementary material online, Figure S1) and increased CSA (+50 ± 38%, *P =0.0101 vs. WT NaCl) with no change in CML (+9 ± 12%, *P =0.3317 vs. WT NaCl). Blood pressure was similar in both genotypes (Supplementary material online, Figure S1). Similar to males, nulli-pari female CKOs developed LV dysfunction in response to Iso (fractional shortening: CKO, 22 ± 6%; WT, 44 ± 5%; #P <0.0001; #P indicates two-tailed Student's t-test). The β1-selective blocker metoprolol largely attenuated Iso-induced adverse effects in CKO mice, while bromocriptine had no effect (Table 2). While Iso stimulation enhanced phosphorylation of troponin I (TnI) at the protein kinase A (PKA) and C (PKC) sites similarly in CKO and WT mice (Figure 1D and E), a more pronounced increase in cardiac fibrosis, cardiomyocyte necrosis, and inflammation and higher expression of atrial and brain natriuretic peptides (BNP) were observed in CKO compared with WT mice (Figure 1F–K; Supplementary material online, Figure S1).
Cardiac function and dimension in wild-type and CKO mice after 7 days of Iso treatment
| NaCl | Iso | |||||
|---|---|---|---|---|---|---|
| WT n = 10 | CKO n = 11 | WT n = 27 | CKO n = 58 | CKO Meto n = 12 | CKO BR n = 8 | |
| LVEDD (mm) | 3.9 ± 0.3 | 4.0 ± 0.4 P > 0.9999a | 4.0 ± 0.5 P > 0.9999b | 4.6 ± 0.4 P < 0.0001b P < 0.0001a | 4.0 ± 0.3 P < 0.0001c | 4.3 ± 0.4 P = 0.0510c |
| LVESD (mm) | 2.4 ± 0.4 | 2.7 ± 0.4 P = 0.5549a | 2.5 ± 0.6 P > 0.9999b | 3.6 ± 0.7 P < 0.0001b P < 0.0001a | 2.5 ± 0.6 P < 0.0001c | 3.3 ± 0.7 P = 0.2600c |
| FS (%) | 39 ± 7 | 34 ± 6 P = 0.4701a | 38 ± 8 P > 0.9999a | 21 ± 11 P = 0.0002b P < 0.0001a | 37 ± 13 P < 0.0001c | 24 ± 10 P = 0.4680c |
| HR (b.p.m.) | 500 ± 127 | 421 ± 135 P = 0.0881a | 544 ± 65 P = 0.3662b | 528 ± 80 P = 0.0008b P = 0.8809a | 534 ± 60 P = 0.8392c | 520 ± 42 P = 0.7832c |
| NaCl | Iso | |||||
|---|---|---|---|---|---|---|
| WT n = 10 | CKO n = 11 | WT n = 27 | CKO n = 58 | CKO Meto n = 12 | CKO BR n = 8 | |
| LVEDD (mm) | 3.9 ± 0.3 | 4.0 ± 0.4 P > 0.9999a | 4.0 ± 0.5 P > 0.9999b | 4.6 ± 0.4 P < 0.0001b P < 0.0001a | 4.0 ± 0.3 P < 0.0001c | 4.3 ± 0.4 P = 0.0510c |
| LVESD (mm) | 2.4 ± 0.4 | 2.7 ± 0.4 P = 0.5549a | 2.5 ± 0.6 P > 0.9999b | 3.6 ± 0.7 P < 0.0001b P < 0.0001a | 2.5 ± 0.6 P < 0.0001c | 3.3 ± 0.7 P = 0.2600c |
| FS (%) | 39 ± 7 | 34 ± 6 P = 0.4701a | 38 ± 8 P > 0.9999a | 21 ± 11 P = 0.0002b P < 0.0001a | 37 ± 13 P < 0.0001c | 24 ± 10 P = 0.4680c |
| HR (b.p.m.) | 500 ± 127 | 421 ± 135 P = 0.0881a | 544 ± 65 P = 0.3662b | 528 ± 80 P = 0.0008b P = 0.8809a | 534 ± 60 P = 0.8392c | 520 ± 42 P = 0.7832c |
LVEDD, LV end-systolic diameter (LVESD), FS (%), and heart rate were determined by echocardiography after 7 days of indicated treatment: Iso (30 mg/kg/day), metroprolol (Meto: 400 mg/kg/day), and bromocriptine (BR: 4 mg/kg/day). Data are depicted as mean ± SD.
aP vs. corresponding WT; P-values were assessed by two-way ANOVA followed by Bonferroni post hoc test.
bP vs. corresponding genotype NaCl.
cP vs. CKO + Iso. P-values were computed by two-tailed Student's t-test.
Cardiac function and dimension in wild-type and CKO mice after 7 days of Iso treatment
| NaCl | Iso | |||||
|---|---|---|---|---|---|---|
| WT n = 10 | CKO n = 11 | WT n = 27 | CKO n = 58 | CKO Meto n = 12 | CKO BR n = 8 | |
| LVEDD (mm) | 3.9 ± 0.3 | 4.0 ± 0.4 P > 0.9999a | 4.0 ± 0.5 P > 0.9999b | 4.6 ± 0.4 P < 0.0001b P < 0.0001a | 4.0 ± 0.3 P < 0.0001c | 4.3 ± 0.4 P = 0.0510c |
| LVESD (mm) | 2.4 ± 0.4 | 2.7 ± 0.4 P = 0.5549a | 2.5 ± 0.6 P > 0.9999b | 3.6 ± 0.7 P < 0.0001b P < 0.0001a | 2.5 ± 0.6 P < 0.0001c | 3.3 ± 0.7 P = 0.2600c |
| FS (%) | 39 ± 7 | 34 ± 6 P = 0.4701a | 38 ± 8 P > 0.9999a | 21 ± 11 P = 0.0002b P < 0.0001a | 37 ± 13 P < 0.0001c | 24 ± 10 P = 0.4680c |
| HR (b.p.m.) | 500 ± 127 | 421 ± 135 P = 0.0881a | 544 ± 65 P = 0.3662b | 528 ± 80 P = 0.0008b P = 0.8809a | 534 ± 60 P = 0.8392c | 520 ± 42 P = 0.7832c |
| NaCl | Iso | |||||
|---|---|---|---|---|---|---|
| WT n = 10 | CKO n = 11 | WT n = 27 | CKO n = 58 | CKO Meto n = 12 | CKO BR n = 8 | |
| LVEDD (mm) | 3.9 ± 0.3 | 4.0 ± 0.4 P > 0.9999a | 4.0 ± 0.5 P > 0.9999b | 4.6 ± 0.4 P < 0.0001b P < 0.0001a | 4.0 ± 0.3 P < 0.0001c | 4.3 ± 0.4 P = 0.0510c |
| LVESD (mm) | 2.4 ± 0.4 | 2.7 ± 0.4 P = 0.5549a | 2.5 ± 0.6 P > 0.9999b | 3.6 ± 0.7 P < 0.0001b P < 0.0001a | 2.5 ± 0.6 P < 0.0001c | 3.3 ± 0.7 P = 0.2600c |
| FS (%) | 39 ± 7 | 34 ± 6 P = 0.4701a | 38 ± 8 P > 0.9999a | 21 ± 11 P = 0.0002b P < 0.0001a | 37 ± 13 P < 0.0001c | 24 ± 10 P = 0.4680c |
| HR (b.p.m.) | 500 ± 127 | 421 ± 135 P = 0.0881a | 544 ± 65 P = 0.3662b | 528 ± 80 P = 0.0008b P = 0.8809a | 534 ± 60 P = 0.8392c | 520 ± 42 P = 0.7832c |
LVEDD, LV end-systolic diameter (LVESD), FS (%), and heart rate were determined by echocardiography after 7 days of indicated treatment: Iso (30 mg/kg/day), metroprolol (Meto: 400 mg/kg/day), and bromocriptine (BR: 4 mg/kg/day). Data are depicted as mean ± SD.
aP vs. corresponding WT; P-values were assessed by two-way ANOVA followed by Bonferroni post hoc test.
bP vs. corresponding genotype NaCl.
cP vs. CKO + Iso. P-values were computed by two-tailed Student's t-test.
Chronic β1-adrenergic receptor stimulation impairs oxidative metabolism in STAT3-deficient hearts
![Cardiac energy deficit in Iso-stimulated CKO mice. Quantification of (A) myocardial perfusion by [11C]acetate positron emission tomography (n = 4–5), (B) O2 consumption by [11C]acetate positron emission tomography (n = 4–5) at baseline and after 3 days of Iso treatment, (C) cardiac adenosine diphosphate content, (D) adenosine triphosphate content (n = 4–5), and (E) adenosine diphosphate to adenosine triphosphate ratio after 3 days of Iso treatment (n = 5–7). (F) Original trace of O2 consumption in isolated mitochondria before and after addition of pyruvate/malate (5/5 mM), adenosine diphosphate (1 mM), and oligomycin (1.2 µM), (G) average values of state 4 (−adenosine diphosphate) and state 3 respiration (+adenosine diphosphate; n = 6–7), and (H) formation of superoxide (O2−; n = 5–7); or (I) H2O2 emission (n = 5) in mitochondria isolated from hearts of untreated or Iso-treated (24 h) mice. (J) Representative western blot of STAT3 protein in isolated mitochondria or left ventricular tissue from wild-type and CKO mice with voltage-dependent anion channel 1 as mitochondrial, glyceraldehyde-3-phosphate dehydrogenase as cytosolic marker and Ponceau S as loading control. Data are mean ± SD. P-values were evaluated by two-way ANOVA and Bonferroni post hoc test (A and B) or by two-tailed Student's t-test (C–E, G–I).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/38/5/10.1093_eurheartj_ehw086/2/m_ehw08602.jpeg?Expires=1747893775&Signature=VQX080d96CHQ4MC~W22coswS-fhTuwVNs6gSsTHNhNBO6DS04a0cWzT3WdF3J3oVdap7u4khMRr5URFx9pzVGfbD02g-AI7rG-GoyjAh9GqwCDNSyu45DGnCRhpAHutBXXnCDgZ3FifahU9SlZW2rHRCjrw6rENjebFMIVvTjJ~Fs2kvCsaJrxMWMxYCUyyyIXYDEDH8pA98sOTj~Xl8VNrATe9jORnpffF0ysC~joMEEwAnDI6--yzYcobgcwV9gjJ2672Mm-Ua5Ph6NZ6abAgTgHPxClikugZinyEW2DfMwLF-2KWJxbQlSQdazn~QRKE-iHT05UlCkKG4QQFzHQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cardiac energy deficit in Iso-stimulated CKO mice. Quantification of (A) myocardial perfusion by [11C]acetate positron emission tomography (n = 4–5), (B) O2 consumption by [11C]acetate positron emission tomography (n = 4–5) at baseline and after 3 days of Iso treatment, (C) cardiac adenosine diphosphate content, (D) adenosine triphosphate content (n = 4–5), and (E) adenosine diphosphate to adenosine triphosphate ratio after 3 days of Iso treatment (n = 5–7). (F) Original trace of O2 consumption in isolated mitochondria before and after addition of pyruvate/malate (5/5 mM), adenosine diphosphate (1 mM), and oligomycin (1.2 µM), (G) average values of state 4 (−adenosine diphosphate) and state 3 respiration (+adenosine diphosphate; n = 6–7), and (H) formation of superoxide (O2−; n = 5–7); or (I) H2O2 emission (n = 5) in mitochondria isolated from hearts of untreated or Iso-treated (24 h) mice. (J) Representative western blot of STAT3 protein in isolated mitochondria or left ventricular tissue from wild-type and CKO mice with voltage-dependent anion channel 1 as mitochondrial, glyceraldehyde-3-phosphate dehydrogenase as cytosolic marker and Ponceau S as loading control. Data are mean ± SD. P-values were evaluated by two-way ANOVA and Bonferroni post hoc test (A and B) or by two-tailed Student's t-test (C–E, G–I).
Chronic β1-adrenergic receptor stimulation impairs mitochondrial pyridine nucleotide recovery and reactive oxygen species elimination in STAT3-deficient cardiomyocytes
Maximal pyruvate/malate (or glutamate/malate-)-supported respiration in the presence of ADP (‘State 3’), O2− formation, and H2O2 emission in mitochondria isolated from untreated or Iso-treated WT and CKO mice were comparable (Figure 2F–I; Supplementary material online, Figure S3), although mitochondrial STAT3 protein content of CKO mice was decreased to a similar extent as in LV tissue (Figure 2J).
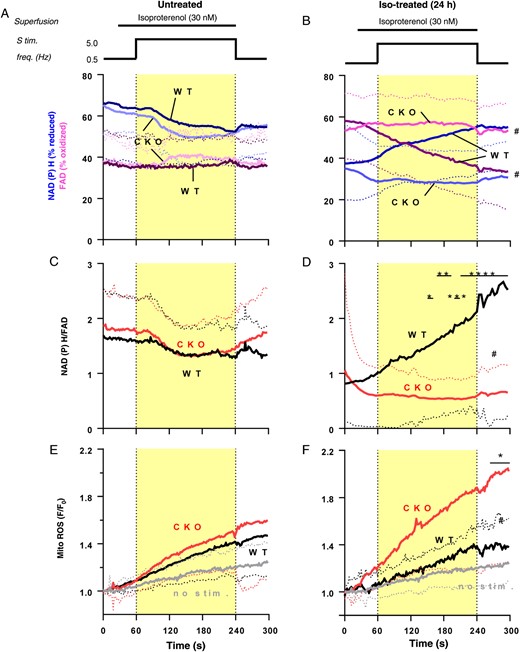
Mitochondrial oxidation and reactive oxygen species production in cardiomyocytes from Iso-treated CKO and wild-type mice. Isolated cardiomyocytes from untreated or Iso-treated (24 h) mice were electrically stimulated (0.5 Hz) and then exposed to Iso (30 nM) and increased stimulation frequency of 5 Hz for 3 min. (A and B) Auto-fluorescence of nicotinamide adenine dinucleotide phosphate (blue traces) and flavin adenine dinucleotide (magenta traces), (C and D) the ratio of nicotinamide adenine dinucleotide phosphate/flavin adenine dinucleotide, and (E and F) mitochondrial .O2− formation indexed by MitoSOX. n = 8–43; data are means ± SD. #P <0.05 CKO vs. wild type, two-way ANOVA; *P <0.05, **P <0.01, ***P <0.001, and ****P <0.0001 wild type vs. CKO Bonferroni post-test of two-way ANOVA.
Chronic β1-adrenergic receptor stimulation impairs substrate utilization in CKO mice
![Analysis of cardiac metabolism in Iso-stimulated CKO and wild-type mice. Quantification of (A) serum free fatty acid levels, (B) myocardial uptake of the palmitate analogue 14(R, S)-[18F]fluoro-6-thia-heptadecanoic acid, in vivo by positron emission tomography, (C) left ventricular triglycerides content, and (D) blood glucose levels in control and Iso-stimulated (3 days) mice (n = 6–7). (E) [18F]fluorodeoxyglucose uptake in cardiomyocytes (isolated from mice after 24 h Iso stimulation in vivo) baseline and with acute insulin (50 nM), (F) or NRG1 stimulation (14 nM) (cardiomyocyte from n = 3–4 mice per group). (G) Pyruvate content in cardiomyocytes isolated from mice exposed for 24 h to Iso stimulation in vivo (cardiomyocyte measurement from n = 3 mice per group). (H) Western blots depicting left ventricular GLUT1 and 4 protein levels (Ponceau S as loading control) and (I) GLUT4 quantification at baseline and after 3 days of Iso stimulation (n = 5–8). (J) Immunohistochemistry for GLUT4 (GLUT4: red; wheat germ agglutinin: green; 4',6-diamidino-2-phenylindole staining: blue) on left ventricular cryosections, (K) Kaplan–Meier survival analysis, and (L) left ventricular function after 7 days of Iso stimulation alone or Iso stimulation with etomoxir in comparison with baseline (etomoxir: 25 mg/kg/day; n = 5–10). Data are mean ± SD. P-values were computed by log-rank (Mantel–Cox) test (J) or two-way ANOVA and Bonferroni post hoc test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/38/5/10.1093_eurheartj_ehw086/2/m_ehw08604.jpeg?Expires=1747893775&Signature=f2kgJlqnZ2f3EksNuGo39wRsmeBMq9j4CV2FsSiGox7ey9YQHnV4yGPSBsXavt7xVrw-XsX2~WAz1kQ68TP7Rr39xilMOnMDidiJ5g3b0j4FFz~ifyAxmzmHyKYF-df3T4nJzOkwzjPTT3L6ulB7uv6zNwB4W6Ospoqr4CM7pl4t9lQU5l-EW~CVZYM-WXA~3pNefMzf~VMhKvIT8KiWDXvCx~cqrPzy0UdGpb1CDaq3Xko68xk9pniqE136v6CTwu8cZIBb99-gmjytlVemIkn5En2tuIKbZRISAQw2Ec3aC5U5DsDKeL2Pols0FNVo93hQAy7D1lAaLN9VrWl3sQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Analysis of cardiac metabolism in Iso-stimulated CKO and wild-type mice. Quantification of (A) serum free fatty acid levels, (B) myocardial uptake of the palmitate analogue 14(R, S)-[18F]fluoro-6-thia-heptadecanoic acid, in vivo by positron emission tomography, (C) left ventricular triglycerides content, and (D) blood glucose levels in control and Iso-stimulated (3 days) mice (n = 6–7). (E) [18F]fluorodeoxyglucose uptake in cardiomyocytes (isolated from mice after 24 h Iso stimulation in vivo) baseline and with acute insulin (50 nM), (F) or NRG1 stimulation (14 nM) (cardiomyocyte from n = 3–4 mice per group). (G) Pyruvate content in cardiomyocytes isolated from mice exposed for 24 h to Iso stimulation in vivo (cardiomyocyte measurement from n = 3 mice per group). (H) Western blots depicting left ventricular GLUT1 and 4 protein levels (Ponceau S as loading control) and (I) GLUT4 quantification at baseline and after 3 days of Iso stimulation (n = 5–8). (J) Immunohistochemistry for GLUT4 (GLUT4: red; wheat germ agglutinin: green; 4',6-diamidino-2-phenylindole staining: blue) on left ventricular cryosections, (K) Kaplan–Meier survival analysis, and (L) left ventricular function after 7 days of Iso stimulation alone or Iso stimulation with etomoxir in comparison with baseline (etomoxir: 25 mg/kg/day; n = 5–10). Data are mean ± SD. P-values were computed by log-rank (Mantel–Cox) test (J) or two-way ANOVA and Bonferroni post hoc test.
Blood glucose levels were similar at baseline and slightly reduced by Iso stimulation in both genotypes (Figure 4D). To rule out any contribution by inflammatory cells or fibroblasts (known for high glucose uptake), isolated cardiomyocytes from WT and CKO mice (untreated or after 24 h in vivo Iso treatment) were used. Baseline and insulin-induced glucose uptake (assayed by [18F]fluorodeoxyglucose, [18F]FDG) were comparable while NRG1-induced glucose uptake was lower in CKO compared with WT cardiomyocytes from untreated mice (Supplementary material online, Figure S4). Glucose uptake (baseline and in response to insulin or NRG1) and pyruvate and lactate content (−64 ± 2%, #P =0.0230) were significantly lower in CKO compared with WT cardiomyocytes from Iso-treated mice (Figure 4E–G).
Pharmacological modulation of glucose uptake impacts on cardiac function and survival of CKO and wild-type mice exposed to chronic β1-adrenergic receptor stimulation
Baseline and Insulin-induced glucose uptake in cardiomyocytes is mainly mediated by GLUT4. Iso treatment decreased the expression and cardiomyocyte membrane localization of GLUT4 in LVs from CKO compared with WT mice, while GLUT1 was not affected (Figure 4H–J). Etomoxir, known to improve glycolysis in part by enhancing GLUT4 activity,10 increased membrane localization of GLUT4 in Iso-stimulated CKO mice, which was associated with improved survival and cardiac function (Figure 4J–L). Also perhexiline enhances GLUT4-mediated glucose uptake5 and improved survival and cardiac function in Iso-stimulated CKO mice (Supplementary material online, Figure S4). Perhexiline increased pyruvate levels in LV tissue of Iso-stimulated CKO mice (Supplementary material online, Figure S4). Conversely, impairing glycolysis with 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, a small-molecule inhibitor of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3,11 provoked high mortality and cardiac dysfunction in Iso-treated WT mice (Supplementary material online, Figure S4).
Cardiac ErbB4 signalling sustains glucose metabolism during chronic β1-adrenergic receptor stimulation
![miR-199a and miR-7a and disturbed NRG1/ErbB and GLUT4 function in STAT3-deficient cardiomyocytes. (A) Western blots for ErbB4 and ErbB2 left ventricular proteins and quantification of (B) ErbB4 (n = 8–12) and (C) ErbB2 (n = 6–8; Ponceau S as loading control). (D) Western blots for p-ADAM17 (phosphorylated at T735) and total ADAM17 protein with (E) ratio of p-ADAM17 to total ADAM17 left ventricular protein in control and in Iso-stimulated (3 days) mice (n = 6–8). (F) Western blot depicting ErbB4 protein levels, (G) [18F]fluorodeoxyglucose uptake after NRG1 (14 nM) or (H) insulin stimulation (50 nM) (I) pyruvate content and (J) metabolic activity by MTS in neonatal rat cardiomyocytes transfected with siRNA against ErbB4 (ErbB4-KD) or scrambled siRNA (control). (K) Western blot showing ErbB4 protein in neonatal rat cardiomyocytes with adenoviral miR-199a overexpression (miR-199a-OE) compared with control adenovirus (left) or in neonatal rat cardiomyocytes with an antagomir-mediated knockdown of miR-199a (miR-199a-KD) and control antagomir (right). (L) Western blot showing ErbB4 protein in neonatal rat cardiomyocytes transfected with pre-miR-7a (miR-7a-OE) and control pre-miR. (M) Western blot showing GLUT4 in neonatal rat cardiomyocytes with adenoviral miR-199a-OE compared with control and (N) in neonatal rat cardiomyocytes with miR-7a-OE compared with control. All data are mean ± SD. P-values were computed by two-way ANOVA and Bonferroni post hoc test (B, C, E, G, and H) or by two-tailed Student's t-test (I and J). Experiments in neonatal rat cardiomyocytes were performed in at least three independent cell isolations in triplicates.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/38/5/10.1093_eurheartj_ehw086/2/m_ehw08605.jpeg?Expires=1747893775&Signature=TreRejA2d4aunMemeBSYxElvLHqpxcVKj4POBf3mlgk33VHVGAZoQx8PsycGMi~FUSbMu8Vjp28eVZOk64LHxHcFJmAF4421kOIqA3VSSpig2BxfOoUnI-B7sHWzUcBjkKpNolzlXezFgm2NtiXYjXveOQAWuItaY8VGpd3Ao7sqOrbwkkN0CRmUfXHjIsus8SFf2qEsS9t5~meLG4F4SVIbDkMunCjGYeHC-RSp4WeDMsJ4E~lwd8b8uXhOss8KsDBmzUts1Vj8ypsRrm44cdaADwPikPA3TRRSepDal0kcoktige7qxJgUHH-u6OknjxP2XRXICtQiQWc~b4Q9oQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
miR-199a and miR-7a and disturbed NRG1/ErbB and GLUT4 function in STAT3-deficient cardiomyocytes. (A) Western blots for ErbB4 and ErbB2 left ventricular proteins and quantification of (B) ErbB4 (n = 8–12) and (C) ErbB2 (n = 6–8; Ponceau S as loading control). (D) Western blots for p-ADAM17 (phosphorylated at T735) and total ADAM17 protein with (E) ratio of p-ADAM17 to total ADAM17 left ventricular protein in control and in Iso-stimulated (3 days) mice (n = 6–8). (F) Western blot depicting ErbB4 protein levels, (G) [18F]fluorodeoxyglucose uptake after NRG1 (14 nM) or (H) insulin stimulation (50 nM) (I) pyruvate content and (J) metabolic activity by MTS in neonatal rat cardiomyocytes transfected with siRNA against ErbB4 (ErbB4-KD) or scrambled siRNA (control). (K) Western blot showing ErbB4 protein in neonatal rat cardiomyocytes with adenoviral miR-199a overexpression (miR-199a-OE) compared with control adenovirus (left) or in neonatal rat cardiomyocytes with an antagomir-mediated knockdown of miR-199a (miR-199a-KD) and control antagomir (right). (L) Western blot showing ErbB4 protein in neonatal rat cardiomyocytes transfected with pre-miR-7a (miR-7a-OE) and control pre-miR. (M) Western blot showing GLUT4 in neonatal rat cardiomyocytes with adenoviral miR-199a-OE compared with control and (N) in neonatal rat cardiomyocytes with miR-7a-OE compared with control. All data are mean ± SD. P-values were computed by two-way ANOVA and Bonferroni post hoc test (B, C, E, G, and H) or by two-tailed Student's t-test (I and J). Experiments in neonatal rat cardiomyocytes were performed in at least three independent cell isolations in triplicates.
ErbB4 knockdown by siRNA in neonatal rat cardiomyocytes (NRCM) (mRNA: −70 ± 3% vs. control, #P =0.0007) blunted NRG1-, but not insulin-induced glucose uptake and reduced pyruvate content and metabolic activity (Figure 5F–J).
Inhibition of ErbB4 signalling with AST-1306 (80 mg/kg/day) had no effect on LV function and survival in control WT mice [LVFS (7 days): DMSO control, 41 ± 8%, n = 8 vs. DMSO/AST, 38 ± 7%, n = 7, *P >0.9999] but increased mortality and LV dysfunction when combined with Iso treatment [LVFS (7 days): DMSO/Iso, 30 ± 7%, n = 8 vs. DMSO/AST/Iso, 15 ± 13%, n = 4, *P =0.0013; 2 of 6 DMSO/AST/Iso-stimulated mice died within 7 days].
microRNA-199a and microRNA-7a are involved in STAT3-dependent regulation of glucose uptake in the heart
Since Iso did not regulate STAT3 activation (phosphorylation of tyrosine-705 or serine-727) in NRCM directly (data not shown), we evaluated whether miR-199a, which is suppressed by STAT3,16 would target ErbB4 and/or GLUT4. In NRCM, miR-199a overexpression decreased while miR-199a reduction increased ErbB4 protein levels (Figure 5K). An anti-Argonaute ribonucleoprotein immunoprecipitation (RIP) assay confirmed direct regulation of ErbB4 by miR-199a (Supplementary material online, Results). MiR-199a had no effect on GLUT4 (Figure 5M). In silico analyses using TargetScan and RNA22 identified GLUT4 as a target of miR-7a in human, rat, and/or mouse. MiR-7a expression was similar at baseline in both genotypes (*P >0.9999) but increased in CKO compared with WT after Iso treatment (1.8-fold after 3 days, *P =0.0003). MiR-7a overexpression did not alter ErbB4 but decreased GLUT4 protein levels (miR-7a-KD: −36 ± 12%, #P =0.0208, Figure 5L and N).
Proof-of-concept analyses in left ventricular tissue from peripartum cardiomyopathy patients
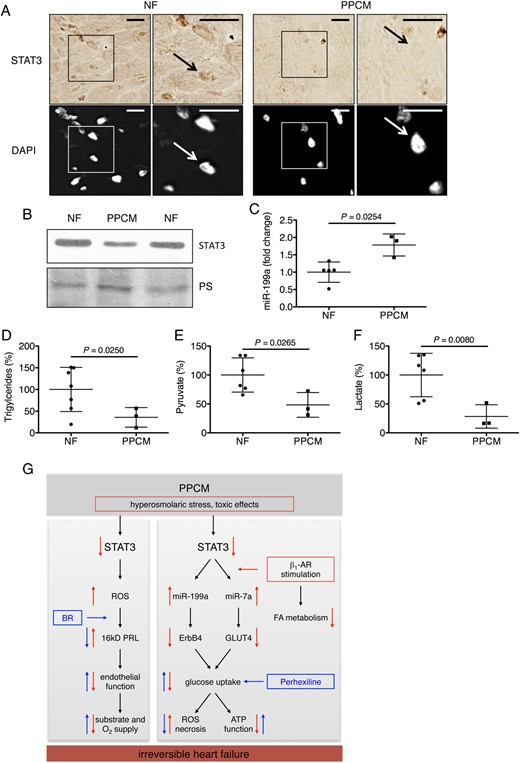
Analyses in left ventricular tissue from peripartum cardiomyopathy patients and non-failing controls. (A) Immunohistochemistry (B) and western blot depicting STAT3 protein levels, (C) miR-199a expression, (D) triglycerides, (E) pyruvate, and (F) lactate content in LV tissue of peripartum cardiomyopathy hearts (n = 3) and LV tissue from non-failing hearts (n = 5–7). (G) Scheme depicting role of STAT3-dependent signalling networks for adverse effects by β1-adrenergic receptor stimulation in peripartum cardiomyopathy. Data are mean ± SD. P-values were computed by two-tailed Student's t-test.
There is evidence that hyperosmolar conditions could be associated with the development of PPCM.17 Hyperosmolar conditions reduce STAT3 expression,18 which we observed also in NRCM (Supplementary material online, Figure S5).
Discussion
Our study suggests that β1-AR stimulation in PPCM patients with acute HF is associated with adverse outcome, since in the German registry,7 all PPCM patients treated with dobutamine needed either heart transplantation or an LVAD while almost all PPCM patients in comparably critical conditions but without dobutamine treatment recovered. A possible explanation why dobutamine may promote cardiac damage and HF progression could be the frequently observed low cardiac STAT3 expression in PPCM patients,3 also confirmed here in some patients of the Dob-group. Indeed, the β-AR agonist Iso worsened HF in our experimental model for PPCM, i.e. postpartum CKO mice. This is independent from the cleaved 16 kDa-PRL, since HF could not be prevented by bromocriptine. Iso induced HF and high mortality in non-pregnant CKO mice, which could be attenuated by the β1-AR blocker metoprolol, indicating that STAT3 deficiency alone is sufficient for β1-AR-activation-induced terminal HF. One possible mechanism for reduced cardiac STAT3 expression in PPCM could be hyperosmolar stress, which decreases STAT3 in cardiomyocytes. Indeed, delivery itself, in particular when associated with substantial bleeding, produces hypovolemic and therefore hyperosmolar conditions. Furthermore, an ethnic tradition in Nigeria is to treat postpartum women with a high-salt diet, which is likely to generate hyperosmolarity. These women have the highest incidence of PPCM (1:100).17 Additional mechanisms may involve previous anthracyline treatment that decreases cardiomyocyte STAT319 and that is a risk factor for PPCM.7,20
The link between STAT3 deficiency and toxic effects of β1-AR stimulation involves metabolic impairments. Iso depletes serum FFA, cardiac FFA uptake, and triglycerides as previously shown in rats21 and confirmed in Iso-treated WT and CKO mice. As a consequence, glycolysis becomes the predominant metabolic pathway as confirmed by the observation that attenuating glycolysis during Iso stimulation caused cardiac dysfunction and high mortality in WT mice while improving glucose uptake by etomoxir or perhexiline5,10 improved cardiac function and survival in Iso-treated CKO mice. The glycolytic products lactate and pyruvate were reduced in STAT3-deficient cardiomyocytes from Iso-treated CKO mice as the consequence of impaired glucose uptake and possibly by additional impairment in rate limiting glycolytic enzymes. Subsequently, the Krebs cycle-mediated regeneration of the mitochondrial pyridine pool (NADH, NADPH, and FADH2) that fuels the ETC for ATP production was lower in STAT3-deficient cardiomyocytes from Iso-treated CKO mice thereby explaining their lower cardiac energy status and possibly decreased cardiac function. The Krebs cycle also regenerates NADPH, which is required to detoxify ROS.22,23 Therefore, the impaired NADPH regeneration in STAT3-deficient cardiomyocytes was associated with increased mitochondrial ROS formation potentially underlying the induction of necrosis and cardiac damage23 in Iso-stimulated CKO mice. Thus, STAT3 is needed to maintain glucose uptake and subsequent metabolic integrity during β1-AR activation, which prevents cardiac dysfunction and damage.
We could show that STAT3 is required to maintain GLUT4-mediated glucose uptake in cardiomyocytes during Iso stimulation. The role of STAT3 hereby is to suppress Iso-induced up-regulation of miR-7a, a microRNA that targets GLUT4 because (i) reduction of GLUT4 protein was not paralleled by a similar decrease in mRNA, (ii) overexpression of miR-7a reduced GLUT4 protein in cardiomyocytes, and (iii) miR-7a is up-regulated in Iso-stimulated CKO compared with WT mice. Since GLUT4 is also regulated by lysosomal degradation,24,25 additional mechanisms such as impaired GLUT4 recycling and increased degradation may also contribute to the rapid decline of GLUT4 protein in Iso-treated CKO mice and will be evaluated in future studies.
We discovered that a regulatory network involving STAT3, miR-199a, and ErbB4 is important for cardiomyocyte glucose uptake at baseline and during Iso stimulation since (i) NRG1-induced glucose uptake is impaired in control and Iso-treated CKO cardiomyocytes, (ii) pharmacological blockade of ErbB4 by AST-1306 caused HF and increased mortality in Iso-stimulated WT mice, and (iii) Iso activates ErbB4 signalling via ADAM17. Finally, miR-199a, which is suppressed by STAT3,16 directly down-regulates ErbB4 in cardiomyocytes.
We present evidence that this novel STAT3-dependent network is also impaired in dobutamine-treated PPCM patients because they display lower cardiac STAT3, triglyceride, pyruvate and lactate content, and increased miR-199a expression.
In conclusion, while STAT3 deficiency may be causative for PPCM by impairing the cardiac microvascular system and thereby inducing a substrate and O2 shortage, the present study shows that it may also render PPCM patients vulnerable to β1-AR agonist treatment (Figure 6G). This vulnerability is explained by deterioration of STAT3-dependent network control of glucose metabolism that eventually results in the suppression of substrate supply to cardiac mitochondria during β1-AR stimulation, inducing energetic deficit, oxidative stress, cardiomyocyte death, and development of irreversible HF (Figure 6G). Finally, these data discourage the use of dobutamine during acute HF in PPCM patients. Should they nevertheless require dobutamine, they may benefit from improving glucose uptake by perhexiline, a drug already in clinical testing6 (Figure 6G).
Limitations of the study are the retrospective analysis of dobutamine treatment in PPCM patients and the use of Iso and not dobutamine in mice.
Supplementary material
Supplementary material is available at European Heart Journal online.
Authors' contributions
B.S., C.M., J.T., A.R., and A.H. performed statistical analysis. D.H.-K. and C.M. handled funding and supervision. B.S., M.K., M.R.-H., A.H., S.E., J.M.U.S., A.R., A.S., I.T., A.G.N., J.A.S., I.G., S.P., M.M., I.B., R.K., M.J., J.T., M.S., C.H., C.M., and D.H.-K. acquired the data. D.H.-K., C.M., J.K., F.B., and J.B. conceived and designed the research. D.H.-K., C.M., J.B., and F.B. drafted the manuscript. B.S., M.K., M.R.-H., A.H., J.M.U.S., A.R., A.S., A.G.N., I.G., S.P., M.M., M.J., J.T., M.S., F.B., J.B., C.M., and D.H.-K. made critical revision of the manuscript for key intellectual content.
Funding
This study was supported by the Deutsche Forschungsgemeinschaft (Hi 842/9-1 to D. H.-K.; Heisenberg Programm and SFB 894 to C.M., BO3643/3-1 to I.B.) Rebirth II (to D. H.-K.), Nieders. Volkswagenstiftung (to D. H.-K.), and HOMFOR (to C. M.) and conducted within the Finnish Centre of Excellence in Cardiovascular and Metabolic Diseases supported by the Academy of Finland, University of Turku, Turku University Hospital and Åbo Akademi University. Funding to pay the Open Access publication charges for this article was provided by the Medical School Hannover.
Conflict of interest: none declared.
Acknowledgements
We thank Birgit Brandt, Lea Greune, and Silvia Gutzke for technical assistance. Positron emission tomography imaging of [18F]FTHA and [11C]acetate was conducted within the Finnish Centre of Excellence in Cardiovascular and Metabolic Diseases supported by the Academy of Finland, University of Turku, Turku University Hospital and Åbo Akademi University. Heidi Liljenbäck and Mia Ståhle are acknowledged for their help in PET/CT studies.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}