





Abstract
Neutrophil extracellular traps (NETs) are chromatin filaments released by activated polymorphonuclear neutrophils (PMNs) and decorated with granule proteins with various properties. Several lines of evidence implicate NETs in thrombosis. The functional significance and the in vivo relevance of NETs during atherothrombosis in humans have not been addressed until now.
Selective sampling of thrombotic material and surrounding blood from the infarct-related coronary artery (IRA) and the non-IRA was performed during primary percutaneous revascularization in 18 patients with ST-segment elevation acute myocardial infarction (STEMI). Thrombi isolated from IRA contained PMNs and NETs decorated with tissue factor (TF). Although TF was expressed intracellularly in circulating PMNs of STEMI patients, active TF was specifically exposed by NETs obtained from the site of plaque rupture. Treatment of NET structures with DNase I abolished TF functionality measurement. In vitro treatment of control PMNs with plasma obtained from IRA and non-IRA was further shown to induce intracellular up-regulation of TF but not NET formation. A second step consisting of the interaction between PMNs and thrombin-activated platelets was required for NET generation and subsequent TF exposure.
The interaction of thrombin-activated platelets with PMNs at the site of plaque rupture during acute STEMI results in local NET formation and delivery of active TF. The notion that NETs represent a mechanism by which PMNs release thrombogenic signals during atherothrombosis may offer novel therapeutic targets.
Neutrophils are involved in the pathophysiology of infracted coronary arteries in STEMI via NET structures. Platelets, activated by thrombin, are required for NET formation, while the integrity of NET scaffold contributes to the functionality of NET-bound TF. The blockage of NET formation or local neutralization of NET-mediated TF signalling constitutes candidate therapeutic targets.
Introduction
Atherosclerotic plaque rupture in epicardial coronary arteries and subsequent thrombus formation is a localized, catastrophic event that leads to vessel occlusion in cases of acute ST-segment elevation myocardial infarction (STEMI). Tissue factor (TF), the major in vivo initiator of coagulation,1 is critical in arterial thrombosis.2 In this regard, the presence of circulating or blood-borne TF, derived from microparticles, monocytes, and myeloid cells has been reported.3
The role of polymorphonuclear neutrophils (PMNs) in thrombosis is supported by studies showing significant attenuation of thrombus formation in neutropenic mice.2,4 However, the ability of neutrophils to trigger in vivo thrombotic events via production of TF, is a matter of debate.5–7
Recently, the ability of PMNs to generate Neutrophil Extracellular Traps (NETs) which contain extracellular chromatin networks ‘decorated’ with neutrophil granular and cytoplasmic proteins,8 offered a novel view regarding their role in thromboinflammation. Neutrophils and NETs have been linked to thrombosis via various mediators of thrombotic process.9 The ability of PMNs to expose functional TF on NETs7,10,11 identifies them as a source of TF in the interaction between innate immunity, inflammation, and coagulation.2,4,12,13 Although platelets are involved in the formation of NETs14 and are abundantly present in arterial thrombi, their link through PMNs in arterial atherothrombosis is still under investigation.
We show here for the first time that PMNs recruited at the site of plaque rupture during STEMI, become activated following platelet–neutrophil interactions and release NETs decorated with functional TF. TF functionality is dependent on the presence of intact NET structures. NET release in STEMI appears to be a two-step phenomenon in which initial intracellular TF up-regulation in PMNs is followed by the generation of TF-bearing NETs.
Methods
Patients
Patients with STEMI (n = 18) eligible for primary percutaneous revascularization and individuals with normal coronary angiogram on diagnostic coronary angiography used as controls (n = 13), were recruited in the present study. Patients on cardiogenic shock, on chronic dual antiplatelet therapy, history of angina or previous STEMI, with critical stenosis in other coronary arteries and blood disorders, severe hepatic or renal dysfunction, diabetes mellitus, as well as ongoing chronic inflammatory, autoimmune, or malignant diseases were excluded. Patients were transferred immediately at the catheterization laboratory and received unfractionated heparin and aspirin plus ticagrelor oral loading. The study protocol was in accordance with the Declaration of Helsinki and was approved by the Ethics Review Board of the University Hospital of Alexandroupolis. Written informed consent was obtained from each individual.
Selective sampling from the infarct-related coronary artery (IRA) was performed with a dedicated catheter (Export® AP Aspiration Catheter, Medronic Vascular, Danvers, MA, USA). Blood from the non-infarct-related (non-culprit) healthy vessels of STEMI patients (non-IRA) and from control individuals was obtained after coronary ostium engagement, through 6F diagnostic coronary catheters during primary PCI or diagnostic catheterization, respectively. Blood from the IRA, the non-IRA of STEMI patients, and from coronary arteries of control individuals was collected and used for isolation of PMNs, platelets, and plasma within 30–45 min after the transfer to the catheterization laboratory. The mode of sample collection did not affect results concerning NET generation.
Clinical characteristics of patients and conditions of sampling are described in detail in Supplementary material online, Methods and Supplementary Data.
Experimental procedures
Mediators and conditions used for stimulation/inhibition studies, immunofluorescence and FACS experiments, NET structures and protein isolation, MPO/DNA and thrombin-antithrombin (TAT) complex ELISAs, immunoblotting and mRNA analyses are described in detail in Supplementary material online.
Statistical analysis
Data are presented as box charts (means ± SD). Statistical analyses were performed using one-way ANOVA (Scheffé test in uniform n and least significant difference test in non-uniform n for post-hoc comparisons). All statistical analyses were performed with OriginPro 8. P-values <0.05 were considered significant.
Results
Neutrophil extracellular traps are accumulated in infarct-related coronary arteries of patients with ST-segment elevation acute myocardial infarction
We observed ex vivo PMNs releasing NETs isolated from blood samples obtained by IRA aspiration in STEMI patients (n = 18); significant differences were observed compared with non-infarct-related coronary arteries (non-IRA) from the same patients (n = 18), and to control individuals (controls; n = 13), as demonstrated by confocal microscopy [myeloperoxidase/neutrophil elastase (MPO/NE) immunofluorescense, Figure 1 A and B], extracellular DNA levels (Figure 1C), and MPO/DNA complex ELISA in isolated NET structures (Figure 1D). The NET-specific marker MPO/DNA complex was also elevated in plasma samples obtained from IRA compared with non-IRA or controls (Figure 1E), and it was not correlated with TIMI flow grade (see Supplementary material online, Figure S2).
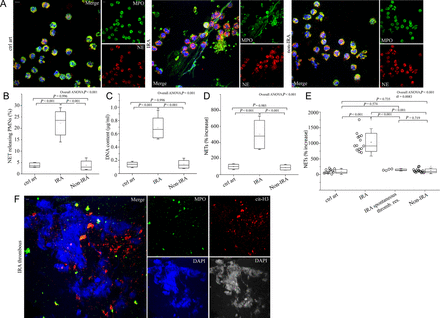
Polymorphonuclear neutrophils of patients with ST-segment elevation acute myocardial infarction release neutrophil extracellular traps at the site of plaque rupture. (A) Neutrophil extracellular traps of polymorphonuclear neutrophils obtained from infarct-related coronary artery, non-infarct-related coronary artery, and control individual coronary arteries/ctrl art (confocal microscopy). (B) Percentage of neutrophil extracellular trap-releasing polymorphonuclear neutrophils, (C) extracellular deoxyribonucleic acid levels, (D) myeloperoxidase–deoxyribonucleic acid complex in isolated neutrophil extracellular trap structures obtained by selective coronary sampling. (E) Myeloperoxidase–deoxyribonucleic acid complex levels in plasma samples obtained from coronary arteries of ST-segment elevation acute myocardial infarction patients and control individuals (least significant difference test was used only in E). (F) Neutrophil extracellular traps visualized in thrombus specimens from patients with ST-segment elevation acute myocardial infarction as extracellular structures decorated with myeloperoxidase and citrulinated H3 (confocal microscopy). Green: myeloperoxidase; red: (A) NE, (F) cit-H3; blue: 4′,6-diamidino-2-phenylindole/deoxyribonucleic acid. One representative out of six (A) or nine (F) independent experiments is shown. Original magnification: (A) ×600, (F) ×400. Scale bar: 5 μm. Box edges for chart boxes of all figures demonstrate mean ± SD. For (B), (C), (D), n = 6.
Accordingly, thrombi contained high numbers of neutrophils either in the form of dense neutrophilic islets (more than 40% coverage of the thrombus, Supplementary material online, Figure S3A) or areas with extensive presence of NETs, (Figure 1F, Supplementary material online, Figure S3B, n = 9).
In patients with STEMI who had undergone spontaneous thrombus resolution (n = 4), MPO/DNA complex levels in plasma from IRA were found significantly lower compared with patients who formed stable thrombi (Figure 1E, n = 14). Furthermore, residual thrombotic material aspirated from patients who underwent spontaneous thrombus resolution demonstrated almost absence of NETs and very low numbers of intact neutrophils (see Supplementary material online, Figure S3A and B, n = 4).
The local NET release in the IRA of STEMI patients points to them as a possible key player in the pathogenesis of the disease.
Intracellular tissue factor in polymorphonuclear neutrophils of ST-segment elevation acute myocardial infarction patients is not always associated with neutrophil extracellular traps
Considering the key role of TF in thrombosis, we investigated the presence of TF on NETs from patients with STEMI. NETs generated ex vivo from PMNs isolated from IRAs were decorated with TF (Figure 2A and B). Furthermore, thrombus specimens demonstrated PMNs and NETs with TF localization, as observed by TF/NE co-localization in confocal microscopy, whereas residual thrombotic material from patients who underwent spontaneous thrombus resolution was characterized by almost complete absence of TF expressing sites (Figure 2C).
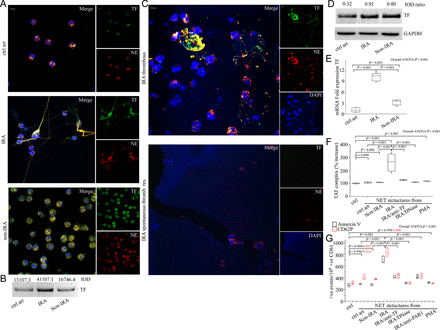
Functional tissue factor is localized in neutrophil extracellular traps generated in infarct-related coronary artery of patients with ST-segment elevation acute myocardial infarction. (A) Localization of tissue factor in polymorphonuclear neutrophils obtained after selective coronary sampling, (confocal microscopy). (B) Tissue factor immunoblotting and integrated optical density of neutrophil extracellular trap proteins. (C) Tissue factor on neutrophil extracellular traps in thrombi specimens from patients with ST-segment elevation acute myocardial infarction, is co-localized with NE extracellularly, (confocal microscopy). Green: tissue factor; red: NE; blue: 4′,6-diamidino-2-phenylindole/deoxyribonucleic acid. One representative out of six (A) or nine (C) independent experiments is shown. Original magnification: ×600. Scale bar: 5 μm. (D) Tissue factor immunoblotting and integrated optical density ratio of tissue factor to glyceraldehyde 3-phosphate dehydrogenase in polymorphonuclear neutrophil lysates. (B), (D) One representative out of three independent experiments is shown. (E) Tissue factor messenger ribonucleic acid expression in polymorphonuclear neutrophils (n = 6) obtained by selective coronary sampling. (F) Thrombin levels in control plasma incubated with isolated neutrophil extracellular trap structures from polymorphonuclear neutrophils obtained by selective coronary sampling. Neutralizing anti-tissue factor antibody was used to inhibit tissue factor-mediated thrombin generation. DNase was used for neutrophil extracellular trap scaffold degradation. Phorbol myristate acetate-induced neutrophil extracellular trap structures in control polymorphonuclear neutrophils were used as negative control. (G) Fluorescence-activated cell-sorting analysis of Annexin V and CD62P in control platelets stimulated with control plasma pre-treated with isolated neutrophil extracellular trap structures from polymorphonuclear neutrophils obtained by selective coronary sampling. FLLRN peptide was used as a PAR-1 antagonist. For (F) and (G) n = 4.
Although NET generation appeared to be a localized phenomenon observed exclusively in the IRA, we observed intracellular TF up-regulation in intact PMNs isolated from both IRA and non-IRA of STEMI patients but not from control individuals, as assessed by immunofluorescence (Figure 2A) and immunoblotting of neutrophil lysates (Figure 2D) and flow cytometry (see Supplementary material online, Figure S4). The observed up-regulation of TF mRNA levels followed a pattern similar to that of protein levels, indicating significant increase of TF expression in PMNs from both IRA and non-IRA of STEMI patients compared with control individuals (Figure 2E).
These findings suggest that although circulating PMNs of patients with STEMI express intracellular TF, its local exposure in IRA is mediated by NETs.
Tissue factor expressed on neutrophil extracellular traps from infarct-related coronary artery needs the deoxyribonucleic acid scaffold to activate the coagulation cascade
TF-thrombin axis has a dual functional role; thrombus formation and signalling via PARs. To study the prothrombotic activity of TF expressed on NETs at the sites of plaque rupture, thrombin levels were analysed with TAT complex ELISA. We observed increased TAT levels after incubation of control plasma with NETs isolated from IRA (Figure 2F). However, TAT levels were normal (Figure 2F) after incubation of control plasma with either NETs isolated from non-IRA or healthy individuals or ‘vacant of TF’-NETs (see Supplementary material online, Figure S5) generated in vitro by phorbol myristate acetate (PMA)-stimulated control PMNs. The increase in thrombin levels was TF-dependent as demonstrated by inhibition with specific neutralizing anti-TF antibody (Figure 2F).
We next assessed the effects of TF-bearing NETs on control platelet activation via PAR signalling. Incubation of control platelets with control plasma pre-treated with IRA-derived NET structures exhibited more pronounced expression of activation markers (Annexin V, CD62P), compared with platelets incubated with control plasma pre-treated with NET structures obtained from non-IRAs or control individuals (Figure 2G). The observed up-regulation of platelet activation markers was TF -dependent as demonstrated by inhibition with specific neutralizing anti-TF antibody (Figure 2G). Moreover, treatment with a PAR-1 antagonist blunted the effect on platelets, demonstrating that NET-bound TF is also able to induce PAR-1 activation, through thrombin generation (Figure 2G).
Prompted by reports in which DNase I interfered in the function of protein components of NETs4,15,16 and considering its therapeutic applications in NET-associated diseases,17,18 we tested whether DNase I affects the functionality of NET-bound TF. Treatment with DNase I abolished thrombin generation in control plasma (Figure 2F) and further platelet activation (Figure 2G).
These data indicate that NETs expose functional TF, able to induce both thrombin generation and platelet activation. NET structure integrity appears to be involved in measurements of TF functionality.
Plasma from infarct-related coronary artery is able to induce tissue factor in vitro but not neutrophil extracellular traps
We assessed the effect of IRA- and non-IRA-obtained plasma on PMN TF expression and NET release in vitro. Control PMNs treated with plasma from IRA or non-IRA demonstrated a similar pattern of intracellular TF increase in both mRNA and protein level in contrast to treatment with plasma from control individuals that showed no TF up-regulation, as observed by immunofluorescence, real time qRT–PCR, immunoblotting (Figure 3A–C), and flow cytometry (Figure 3D). However, no significant increase of NET generation was observed in conditions similar to all previous experiments (Figure 3A and E).
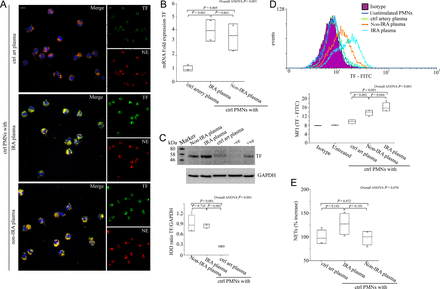
The microenvironment in infarct-related coronary artery induces tissue factor expression but not neutrophil extracellular trap generation in polymorphonuclear neutrophils. (A) Neutrophil extracellular trap generation in control polymorphonuclear neutrophils treated with infarct-related coronary artery plasma, non-infarct-related coronary artery plasma, and plasma obtained from control individuals (confocal microscopy). Green: tissue factor; red: NE; blue: 4′,6-diamidino-2-phenylindole/deoxyribonucleic acid. One representative out of four independent experiments is shown. Original magnification: ×600. Scale bar: 5 μm. (B) Tissue factor mRNA, (C) tissue factor immunoblotting and integrated optical density according to glyceraldehyde 3-phosphate dehydrogenase, and (D) fluorescence-activated cell-sorting analysis with relative MFIs in control polymorphonuclear neutrophils treated with plasma obtained by selective coronary sampling. (C) Unstimulated polymorphonuclear neutrophils and lipopolysaccharide-stimulated peripheral blood mononuclear cells were used as negative and positive controls for tissue factor, respectively. One representative out of four independent experiments is shown. (E) Myeloperoxidase–deoxyribonucleic acid complex in isolated neutrophil extracellular trap structures from control polymorphonuclear neutrophils treated with plasma obtained by selective coronary sampling. For (B), (C), (D), (E) n = 4.
These data indicate that although TF-bearing NETs are formed in the vicinity of plaque rupture, intracellular up-regulation of TF in neutrophils is a generalized phenomenon in patients with STEMI.
Platelets activated in the environment of infarct-related coronary artery are able to induce neutrophil extracellular trap formation
Based on the ability of activated platelets to induce NET release in other disease states,14 we investigated whether activated platelets from the IRA of STEMI patients are able to induce NETs. First, platelets from IRA demonstrated increased levels of surface activation markers compared with platelets from non-IRA or control individuals, as observed by Annexin V and CD62P flow cytometry analysis (Figure 4A and B, Supplementary material online, Figure S6A and B). Moreover, increased number of platelet/neutrophil aggregates (CD61/CD66b double-positive events) was observed in PMNs isolated from IRA compared with non-IRA or control individuals (Figure 4C).
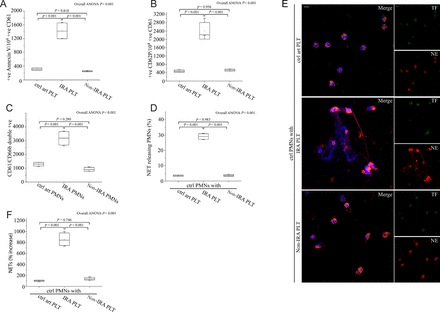
Activated platelets from infarct-related coronary artery are responsible for neutrophil extracellular trap generation. Fluorescence-activated cell-sorting analysis of Annexin V (A) and CD62P (B) on infarct-related coronary artery platelets, non-infarct-related coronary artery platelets and control individual coronary artery platelets. (C) Platelet/polymorphonuclear neutrophil aggregates observed as double-positive CD61/CD66b per 10 000 CD66b positive events with fluorescence-activated cell-sorting analysis in polymorphonuclear neutrophils isolated after selective coronary sampling. (D) Percentage of neutrophil extracellular trap-releasing polymorphonuclear neutrophils and (E) Neutrophil extracellular trap generation by control polymorphonuclear neutrophils treated with platelets isolated after selective coronary sampling, (confocal microscopy). Green: tissue factor; red: NE; blue: 4′,6-diamidino-2-phenylindole/deoxyribonucleic acid. Original magnification: ×600. Scale bar: 5 μm. (F) myeloperoxidase–deoxyribonucleic acid complex in isolated neutrophil extracellular trap structures of control polymorphonuclear neutrophils treated with platelets obtained by coronary sampling. (A), (B), (C), (D), (F) n = 4. (E) One representative out of four independent experiments is shown.
These data indicate activation of platelets at the site of plaque rupture and suggest increased interaction locally with PMNs.
We then tested whether activated platelets play a role in NET generation in the IRA. Control PMNs treated with IRA-derived platelets showed increased NET release (Figure 4D–F), while treatment of PMNs with platelets derived from non-IRA or control individuals did not induce NET formation. Interestingly, NETs generated by activated platelets in the absence of additional IRA-derived plasma demonstrated absence of TF exposure (Figure 4E).
We further observed that IRA-derived plasma was able to activate control platelets in vitro in contrast to non-IRA-derived or control plasma (Figure 5A and B and Supplementary material online, Figure S6A and B). Similar to IRA-derived platelets, in vitro-activated platelets induced the generation of TF-free NETs from control PMNs (Figure 5C–E).
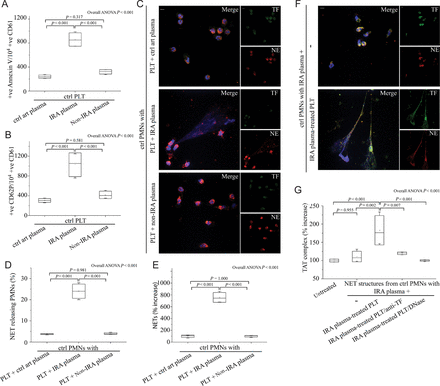
The microenvironment in infarct-related coronary artery activates platelets for subsequent neutrophil extracellular trap generation in polymorphonuclear neutrophils. Fluorescence-activated cell-sorting analysis of Annexin V (A) and CD62P (B) on control platelets treated with plasma obtained by selective coronary sampling. (C), (D), (E) Neutrophil extracellular trap formation by control polymorphonuclear neutrophils incubated with control platelets pre-treated with plasma obtained by selective coronary sampling. (C), (D) Confocal microscopy and (E) myeloperoxidase–deoxyribonucleic acid complex on neutrophil extracellular trap structures. (F) Neutrophil extracellular trap generation after stimulation of control polymorphonuclear neutrophils with plasma from infarct-related coronary artery of ST-segment elevation acute myocardial infarction patients and control platelets pre-treated with infarct-related coronary artery plasma (confocal microscopy). (G) Thrombin levels in control plasma incubated with isolated neutrophil extracellular trap structures from control polymorphonuclear neutrophils stimulated as in (F). Anti-tissue factor antibody was used to neutralize tissue factor-mediated thrombin generation. DNase I was used for neutrophil extracellular trap scaffold degradation. (C) and (F) Green: tissue factor; red: NE; blue: 4′,6-diamidino-2-phenylindole/deoxyribonucleic acid. One representative out of four independent experiments is shown. Original magnification: ×600. Scale bar: 5 μm. For (A), (B), (D), (E), (G) n = 4.
In view of the above findings, the complementary roles of plasma and platelets in the formation of TF-bearing NETs were further investigated by a two-step experimental procedure. The first step was the intracellular expression of TF in control PMNs after stimulation with plasma obtained from STEMI patients (IRA or non-IRA-derived plasma). The second step was NET generation in these TF-loaded PMNs after adding activated platelets, either IRA-isolated platelets or in vitro-activated platelets as described earlier (Figure 5F). NETs derived from this in vitro procedure were also able to induce thrombin generation in a TF-axis-dependent manner, while dismantling of NET scaffold with DNase I abolished the TF-driven TAT measurement (Figure 5G).
This suggests that the generation of TF-expressing NETs follows a two-step process that requires both circulating inflammatory stimuli present in the plasma of STEMI patients and activated platelets primarily present in the vicinity of ruptured plaque.
Thrombin in plasma from infarct-related coronary artery is responsible for platelet activation and subsequent neutrophil extracellular trap generation
Since thrombin is able to activate platelets through PARs,19 we investigated the involvement of thrombin in the local activation of IRA platelets. First we measured the levels of thrombin in patients with STEMI and in control individuals. Thrombin levels were elevated in IRA-derived plasma compared with non-IRA-derived plasma or plasma obtained from control individuals (Figure 6A). Moreover, platelet activation by IRA-derived plasma was thrombin-depended (which mainly signals through PAR-1), as shown by adding thrombin inhibitors antithrombin III or dabigatran, as well as by specific PAR-1 antagonism (Figure 6B and C, Supplementary material online, Figure S6A and B). Furthermore, thrombin inhibition in IRA plasma also attenuated the subsequent NET release by control PMNs incubated with platelets activated with this plasma (Figure 6D). These data were also reproduced by using recombinant thrombin to activate control platelets (Figure 6B–D), while thrombin was unable to induce NET release in control PMNs by itself (Figure 6D). In addition, thrombin inhibition blocked the in vitro activation of platelets making them unable to induce NETs in control PMNs treated with IRA-derived plasma (Figure 6D) as well as prevented TF externalization as observed by immunoblotting of NET proteins (Figure 6E).
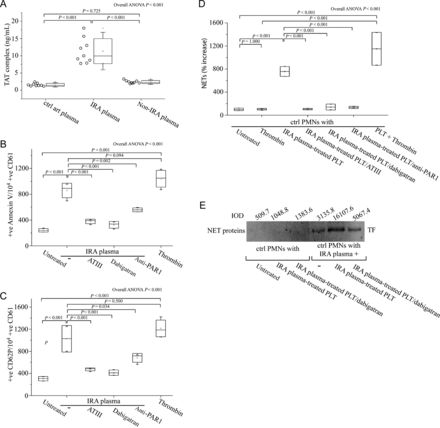
Elevated levels of thrombin in plasma from infarct-related coronary artery are responsible for the local activation of platelets and subsequent neutrophil extracellular trap generation. (A) Thrombin levels in plasma obtained by selective coronary sampling (n = 9, each), as assessed by TAT complex enzyme-linked immunosorbant assay. Fluorescence-activated cell-sorting analysis of Annexin V (B) and CD62P (C) on control platelets treated with plasma from infarct-related coronary artery in the presence or not of thrombin (antithrombin III or dabigatran) and PAR-1 signalling (FLLRN) inhibitors. Recombinant thrombin was used as positive control. (D) Myeloperoxidase–deoxyribonucleic acid complex enzyme-linked immunosorbant assay in neutrophil extracellular traps derived after stimulation of control polymorphonuclear neutrophils with plasma from infarct-related coronary artery of ST-segment elevation acute myocardial infarction patients and control platelets pre-treated with infarct-related coronary artery plasma, in the presence or absence of thrombin inhibitors (antithrombin III or dabigatran). (E) Immunoblotting and integrated optical density of tissue factor in neutrophil extracellular trap proteins derived from in vitro stimulation of control polymorphonuclear neutrophils, as described previously in (D), in the presence or absence of dabigatran. One representative out of four independent experiments is shown. For (B), (C), (D), n = 4.
These data indicate that thrombin in the environment of IRA plasma is a key mediator for the activation of platelets and the ensuing NET generation.
Discussion
Using selective coronary sampling, we could demonstrate for the first time the local accumulation of TF-bearing NETs at the site of coronary thrombosis. We observed blood neutrophils releasing NETs and exposing TF in the IRA but not in the non-IRA of STEMI patients. Moreover, and in agreement with the findings of others,20,21 we observed islets of PMNs and NETs decorated with TF in thrombi obtained from IRAs. The exposed TF on NETs is functional and capable of inducing both thrombin generation and thrombin/PAR1-mediated platelet activation. NET scaffold integrity is necessary for TF functionality and the formation of NETs is driven by thrombin-activated platelets in STEMI.
We showed that the microenvironment in the circulation of STEMI patients promotes the expression of TF by PMNs. We observed the presence of ‘non-NETotic’ TF-loaded PMNs in non-IRA of STEMI patients. Additionally, we were able to reproduce intracellular TF up-regulation in vitro by culturing control PMNs with plasma from both IRA and non-IRA of STEMI patients, but not with plasma from control individuals. We suggest that inflammatory mediator(s) induce neutrophil-derived circulating TF in STEMI patients. Although, various cytokines have been charged as TF inducers,5,11,22 further studies are needed to identify them in STEMI.
The ability of neutrophils to induce in vivo thrombosis by production of TF, is a matter of debate.5–7 However, the discovery of NETs upgraded their prothrombotic role.13 Neutrophilic TF, regardless of its origin, acquired or produced or both, is trafficked on NETs making neutrophils a source of active TF. The neutrophilic source of TF has been recently described;2 the formation of NET locally exposes the TF already present in cells, at the sites of plaque rupture; in contrast the formation of NETs in sepsis is systemic with subsequent dissemination of TF.10 As previous studies have demonstrated the involvement of NETs in thrombosis through further coagulation mediators, such as Factor XII,4 more experiments are required to reveal a possible crosstalk of these molecules with TF on the NET scaffold.
Despite the absence of TF on the surface of PMNs, as assessed by flow cytometry,5 we provide evidence suggesting that the TF exposed on NETs of STEMI patients is functional, able to induce thrombin generation. The generated thrombin can further activate resting platelets through PAR cell signalling, since PAR-1 antagonism attenuated this effect. Accordingly to previous studies suggesting the crucial role of NET integrity in the functionality of proteins epitopes,15 we found that TF-dependent thrombin generation is abolished when NETs are dismantled with DNase I. In accordance, treatment with recombinant human DNase I significantly improves outcome after arterial thrombotic events in mice.23
Activated platelet–neutrophil interactions have been previously shown to induce NET release.14,24 In the present study, we found that activated platelets obtained from IRA and in vitro-activated platelets by IRA-derived plasma are able to induce NET formation in control neutrophils in vitro. The increased thrombin levels found in the IRA and the significant attenuation of platelet-induced NET generation by thrombin inhibitors and PAR blockers suggests a possible role for thrombin in the process. We suggest that activated platelet-PMN interplay continues to generate thrombin, thus supporting thrombogenesis through NETs even after initial thrombus expansion, which limits the ability of plaque-derived TF to migrate in the core of the developing thrombus.25
In conclusion, we show here for the first time the interaction between activated platelets and TF-loaded neutrophils at sites of atherosclerotic plaque rupture for the release of active TF-bearing NETs in humans with acute STEMI (Figure 7). A variety of inflammatory stimuli in STEMI promote the de novo expression of TF in neutrophils. In parallel, locally activated platelets interact with neutrophils for the release of TF-bearing NETs inside the culprit artery. The functionality of TF depends on the integrity of the NET structure and is able to induce thrombin generation and platelet activation, creating a possible vicious cycle that leads to thrombus propagation and stability. The observation that NETs are involved in TF functionality and thrombus stability may trigger further research for novel therapeutic targets.
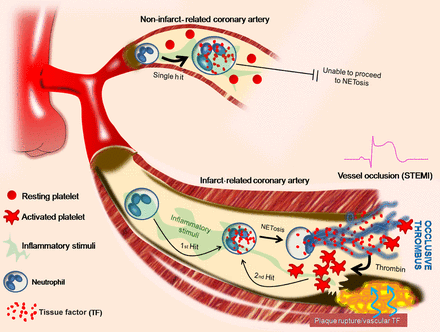
Two ‘hit’ tissue factor/neutrophil extracellular trap model in ST-segment elevation acute myocardial infarction. Schematic representation. Platelets are activated by thrombin in infarct-related coronary artery and interact with polymorphonuclear neutrophils, resulting in neutrophil extracellular traps formation (second step) and exposure of pre-existing tissue factor in polymorphonuclear neutrophils (first step).
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This study was supported by the Hellenic Ministry of Education and General Secretariat for Research and Technology (ESPA SYNERGASIA project/No 898), research grant from the Hellenic Cardiologic Society and National and European Union funds from the ‘Operational Program Education and Life Long Learning’ (NSRF 2007-2013) MIS 379 527. S.K. was supported by the German Federal Ministry of Education and Research (BMBF 01EO1003). Funding to pay the Open Access publication charges for this article was provided by a research grant from the Hellenic Cardiological Society.
Conflict of interest: none declared.
Acknowledgements
The authors would like to thank Dr Akrivi Chrysanthopoulou for her experimental assistance and Prof. Nikolaos Limnios and Dr Vasileios Papadopoulos for their statistical advice. Dr Ashley Goss (Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany) provided dabigatran for our experiments.
References
Author notes
These authors contributed equally.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}