






Abstract
Objective: The use of non-heart-beating donors (NHBD) has been advocated as an alternative to overcome the critical organ shortage in lung transplantation despite the warm ischemic period that may result in primary graft dysfunction. On the contrary, brain death in the heart-beating donor (HBD) may be an underestimated risk factor for donor lung injury. We postulated that 60 min of warm ischemia in the NHBD is less detrimental to the lung than the pathophysiological changes after brain death in the HBD. In this study we compared the quality of lungs from HBD versus NHBD in an isolated reperfusion model. Methods: Pigs (n = 10 per group) were divided into three groups. In group I (HBD), brain death was induced by acute inflation of an intracranial epidural balloon catheter. In group II (CONTROL), the balloon was not inflated. In group III (NHBD), cardiac arrest was induced by myocardial fibrillation. After 5 h of in situ mechanical ventilation, lungs in HBD and CONTROL were preserved with a cold antegrade flush. In NHBD, unflushed grafts were explanted after 1 h of warm ischemia and 4 h of topical cooling in the cadaver. Graft performance was evaluated during 1 h in an isolated ventilation and reperfusion model. Extravascular lung water content (EVLW) was calculated. All data are reported as mean ± SEM. Results: A significant autonomic storm was observed in HBD following balloon inflation. During ex vivo assessment, pulmonary vascular resistance (PVR) at 60 min in HBD (2634 ± 371 dyne s cm−5) was significantly higher as compared with that of CONTROL and NHBD (1894 ± 137 dynes s cm−5 and 1268 ± 111 dynes s cm−5, respectively; p ≪ 0.05). Plateau airway pressure (Plateau AwP) was also higher in HBD (17 ± 1 cmH2O) compared with that of CONTROL (12 ± 1 cmH2O; p ≪ 0.05) and NHBD (14 ± 1 cmH2O; not significant). No significant differences were observed between HBD, CONTROL and NHBD in EVLW (79 ± 1%, 79 ± 0% and 78 ± 1%, respectively), and in PO2/FiO2 (564 ± 58 mmHg, 622 ± 14 mmHg and 578 ± 26 mmHg, respectively). Conclusions: These data indicate that 1-h warm ischemic lungs in our model are less susceptible to ischemia–reperfusion injury than lungs retrieved 5 h after brain death. This study further supports the use of lungs from NHBD for pulmonary transplantation.
1 Introduction
Lung transplantation has become the mainstay of therapy for selected patients suffering from end-stage pulmonary disease to improve survival and to enhance quality of life. The last decade has been marked by a significant increase in transplant procedures and patients on the waiting list [1]. The disparity between a growth in demand and a short supply in donor organs results in longer waiting times for listed patients with a substantial risk of dying prior to transplantation. A number of strategies have evolved in an attempt to deal with this problem of inadequate donor supply. There has been an increasing interest to use organs from circulation-arrested or so-called non-heart-beating donors (NHBD) [2]. There is now an increasing experimental [3] and clinical [4] evidence that pulmonary graft function is not compromised by a limited warm ischemic interval (≪60 min) in the NHBD.
Primary graft dysfunction (PGD), also referred to as ischemia–reperfusion injury, remains a significant cause of early morbidity and mortality in 5% of the recipients, despite refinements in donor care, organ preservation, operative techniques and post-transplant management [5]. PGD can be attributed to a series of harmful events that occur during three phases: the donor phase, the preservation phase and the reperfusion phase. In current brain-dead donors (heart-beating donors, HBD), the pathophysiological events following brain stem herniation may contribute extensively to graft injury prior to retrieval. Both hemodynamic and inflammatory changes following neuro-endocrine disregulation may be responsible for a high risk of acute lung injury. The mechanisms responsible for donor lung injury following brain death, however, are still poorly understood. At the same time, no large animal models resembling the clinical situation are currently available.
A better understanding of these injurious processes may open new perspectives to attenuate donor lung injury following brain death and could support the clinical use of grafts from NHBD. The aim of this study was to compare the impact of explosive brain death in HBD with the warm ischemic damage in NHBD on pulmonary graft function in our isolated porcine lung reperfusion model.
2 Materials and methods
2.1 Animal preparation
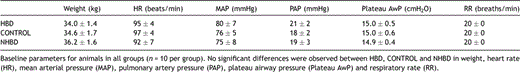
Domestic pigs (n = 30) (weight 35 ± 0.9 kg) (Table 1 ) were chosen as experimental animals, given their availability and resemblance to human physiology. All animals received human care in accordance with the ‘principles of laboratory animal care’ formulated by the National Society of Medical Research and ‘Guide for the care and use of laboratory animals’ published by the National Institutes of Health (NIH Publication No. 86-23, revised 1996).
Baseline parameters in all three study groups
Anesthesia was induced with an intramuscular injection of 2.5 ml zolazepam/tiletamine (Zoletil® 100, Virbac s.a., Carros, France) and 5 ml xylazine (Xyl-M® 2%, V.M.D. nv/sa, Arendonk, Belgium).
Animals were intubated with a 7.5-mm (internal diameter) cuffed endotracheal tube (Hi-Contour™, Mallinckrodt Medical, Athlone, Ireland) and ventilated with a volume-controlled ventilator (Titus®, Dräger, Lübeck, Germany) at a tidal volume of 10 ml/kg body weight with a frequency of 20 breaths/min, a positive end-expiratory pressure (PEEP) of 5 cmH2O and an inspiratory oxygen fraction (FiO2) of 0.5. Respiratory rate (RR) was adjusted to achieve an end-tidal CO2 of 40 mmHg (Table 1). Once baseline values were achieved, ventilatory settings remained unchanged throughout the experimental protocols.
Anesthesia was maintained with 1% isoflurane (Forene®, Abbott laboratories Ltd., Queensborough, Kent, UK) and animals were paralyzed with intermittent boli of 2 mg pancuronium bromide (Pavulon®, N.V. Organon, Oss, The Netherlands).
The neck was explored and great vessels were exposed. A 14 G catheter (Secalon® T, Becton Dickinson Ltd., Singapore) was inserted in the right common carotid artery for hemodynamic monitoring (mean arterial pressure, MAP) and sampling of arterial blood. A 7.5 F pulmonary artery thermodilution catheter (Swan-Ganz, Edwards Lifesciences LLC, Irvine, CA, USA) was inserted through the right external jugular vein to measure pulmonary arterial pressure.
Hemodynamic and ventilatory parameters were continuously recorded.
2.2 Experimental groups
Animals were divided into three groups (n = 10 per group) (Fig. 1 ).

Time line in all three study groups. (a) Balloon inflation; (b) balloon inserted, not inflated; (c) myocardial fibrillation; (d) antegrade flush with cold Perfadex®; (e) explantation without flush; W.I., warm ischemia.
2.2.1 Heart-beating donor group (HBD)
In HBD, explosive brain death was induced.
Animals were placed in prone position, the scalp was incised and the skull exposed. A burr hole was drilled in the right frontoparietal region and an 18 Ch balloon catheter (Foley catheter, Bard Ltd., West Sussex, UK) was positioned in the extradural space pointing caudally. Through a second, contralateral burr hole, a microtransducer (MicroSensor, Codman/Johnson & Johnson, Randolph, MA, USA) was placed in the parenchyma of the left cerebral hemisphere to measure intracranial pressure (ICP). Cranial perfusion pressure (CPP = MAP − ICP) was calculated. The balloon was acutely inflated over 1 min with ±30 ml saline to an ICP of 200 mmHg (time point t = 0 min), resulting in global brain ischemia (CPP ≪ 0 mmHg), brain stem herniation and eventually compression of the medulla oblongata. Brain death was further confirmed by sudden onset of bilateral mydriasis and complete disappearance of brain stem reflexes (cornea and pupillary reflexes).
Animals were ventilated and monitored for 5 h (=in situ observation) with the balloon still inflated and with continuous administration of anesthetic agents. Changes in heart rate (HR), mean arterial pressure, pulmonary artery pressure (PAP) and plateau airway pressure (Plateau AwP) were recorded continuously. Plateau AwP is an optimal estimate of alveolar compliance which is reduced in any form of acute lung injury associated with marked increase in pulmonary membrane permeability [6].
Blood samples were collected before and at different time points after induction of brain death in standard containers with lithium heparin and ethylenediaminetetraacetic acid (EDTA) for analysis of the plasma catecholamines epinephrine and norepinephrine.
After the 5-h observation period, median sternotomy was performed and the pleural cavities and pericardium were opened. The superior and inferior caval veins, ascending aorta and pulmonary trunk were encircled. Heparin (Heparine Leo 5000 IU/ml, LEO Pharma N.V./S.A., Wilrijk, Belgium) was administered (400 IU/kg). The main pulmonary artery was cannulated with a 24 Fr catheter (DLP Inc., Grand Rapids, MI, USA) and secured by a purse-string in the right ventricular outflow tract. The caval veins were ligated, the ascending aorta was clamped. The pulmonary circulation was isolated from the right ventricle by a ligature around the tip of the catheter just distal to the pulmonary valve. The left and right atrial appendage were incised for venting and the lungs were flushed in an antegrade way with 50 ml/kg cold (4 °C) Perfadex® (Vitrolife, Göteborg, Sweden) with the addition of 1 mEq/l trometamol and 60 mg CaCl2. After the flush, catheters were removed, and the heart–lung block was excised and preserved in cold (4 °C) saline for 2 h.
2.2.2 Control group (CONTROL)
Animals were placed in prone position. The balloon catheter was inserted as described in HBD but not inflated (time point t = 0 min). Animals were ventilated and monitored for 5 h (starting at a comparable time point t =0 min with balloon inflation in HBD). CPP remained >30 mmHg. This group served as a sham-operated group to control for mechanical ventilation and surgical interventions in HBD. In situ observation of hemodynamic parameters, airway pressures and plasma catecholamines as well as preservation of the donor organ were performed in an identical way as described in HBD.
2.2.3 Non-heart-beating donor group (NHBD)
In NHBD, sudden cardiac arrest was induced (time point t = 0 min) by induction of myocardial fibrillation with a square pulse generator (amplitude range +15 to −15 V, frequency 50 Hz, current ≪ 300 mA). After sacrifice, the endotracheal tube was disconnected and cadavers were left untouched for 60 min at room temperature (21 °C). This 1-h interval of in situ warm ischemia was followed by a 4-h interval of topical cooling. Therefore, two chest drains (16 Fr Trocar catheter, Argyle, Sherwood Medical, Tullamore, Ireland) were inserted in each pleural space (one deep drain for inflow and one superficial drain for outflow). Cold (6 °C) saline from an ice-cooled reservoir was recirculated through these chest drains with a roller-pump. The superficial outflow drains were connected with an overflow system of 5 cmH2O to ensure that the lungs were well immersed. After 4 h of cold in situ preservation, sternotomy was performed, the heart–lung block was explanted and further stored in cold (4 °C) saline for 2 h.
2.3 Biochemical analysis of plasma catecholamines
The catecholamines epinephrine and norepinephrine were measured in plasma using trace enrichment high performance liquid chromatography with electrochemical detection (HPLC–ED). The catecholamines were extracted off-line using aluminium oxide, and separated on a reversed phase column (MicroSpher 3 μm, C18, 100 mm × 4.6 mm internal diameter, Varian Benelux, Middelburg, The Netherlands). Detection range was from 0 to 2000 ng/l and sensitivity of the method was 0.05 ng/l.
2.4 Preparation of the heart–lung block
After extraction, the grafts in all three groups were prepared in an identical way for evaluation in the isolated reperfusion circuit. The right lung was separated from the heart–lung block and extravascular lung water was determined as described below. The left lung was further prepared during the cold ischemic interval of 2 h for ex vivo evaluation in the isolated reperfusion circuit. A 36 Fr inflow cannula was inserted in the pulmonary trunk through a purse-string on the right ventricular outflow tract. In NHBD, major clots in the pulmonary trunk and left atrium were removed under direct vision. A second cannula for outflow was positioned in the left atrium through the apex of the left ventricle and the mitral valve. Finally, an 8-mm (internal diameter) cuffed endotracheal tube (Hi-Contour™, Mallinckrodt Medical, Athlone, Ireland) was secured in the trachea for ventilation of the graft.
2.5 Preparation of the perfusate
Autologous blood (±600 ml) was rapidly withdrawn from each animal in all three groups at the moment of sacrifice via a central venous catheter and was collected in a sterile bag containing 15,000 IU of heparin. This whole blood was processed with a Cell Saver (Sequestra 1000, Medtronic Inc., Parker, CO, USA) by centrifuging and washing with saline for at least 10 min at 5600 rpm.
White blood cells were removed using a leukocyte filter (Imugard III-RC, Terumo Europe N.V., Haasrode, Belgium). The remaining red blood cell concentrate was then diluted to a haematocrit of 15% with a low potassium dextran solution (Perfadex®) and human albumin (final concentration: 8%). The perfusate was finalized by adding 240 mg/l CaCl2, 10,000 IU/l heparin and 45 ml/l sodium bicarbonate (0.8 M).
2.6 Isolated reperfusion circuit
The reperfusion circuit consisted of a hard-shell reservoir (Vision; GISH biomedical Inc., Irvine, CA, USA), a centrifugal pump (Bio-Medicus, Console 550, Medtronic, Minneapolis, MN, USA) with heater/cooler system (Bio-Cal, Heater Cooler Model 370, Medtronic, Minneapolis, MN, USA) and a membrane gas exchanger with a built in heat exchanger (Vision; GISH biomedical Inc., Irvine, CA, USA). The heating element of the gas exchanger was connected to the heater/cooler.
The left lung was placed in a specially designed evaluation box and mounted in the reperfusion system. The inflow tubing was connected to the cannula in the pulmonary artery. The outflow tubing was connected to the cannula in the left atrium to drain the effluent from the lungs back to the reservoir. Left lungs were ventilated via the endotracheal tube.
Reperfusion of the lungs was started with normothermic (37 °C) oxygenated perfusate after de-airing of the inflow cannula. Pulmonary artery pressure was carefully monitored and allowed to slowly increase to a maximum of 15 mmHg. This resulted in a progressive increase in pulmonary artery flow (PAF) gradually warming up lungs up to 37 °C. Ventilation of both lungs with a FiO2 of 0.5 was not started until the temperature of the pulmonary effluent had reached 34 °C. At that time, the initial oxygen flow (0.4 l/min) supplied to the gas exchanger was switched to a gas mixture of CO2 (8%), O2 (6%) and N2 (86%) thereby obtaining a blood gas value in the influent resembling a normal venous blood sample. Volume-controlled ventilation was also slowly increased to a tidal volume of 140 ml, a frequency of 10 breaths/min, and PEEP of 5 cmH2O.
2.7 Ex vivo functional assessment of pulmonary grafts
Thirty minutes after onset of reperfusion, functional parameters stabilized and were recorded up to 1 h. An electromagnetic flow probe (FF 100T 10-mm probe; Nihon Kohden, Tokyo, Japan) attached to an electromagnetic flow meter (MFV-3200; Nihon Kohden, Tokyo, Japan) was inserted in the tubing on the inflow line for continuous measurement (l/min) of pulmonary artery flow. An 18 G catheter was inserted directly into the main pulmonary artery to measure (mmHg) pulmonary artery pressure. The pressure in the left atrium (LAP), measured on the outflow line, was kept constant at 0 mmHg by adjusting the height of the blood reservoir. Pulmonary vascular resistance (PVR) was calculated using the formula PVR = ((PAP − LAP) × 80)/PAF and expressed in dynes s cm−5. Plateau airway pressure was recorded (cmH2O). PO2, PCO2, pH, saturation and HCO3− were continuously measured in the perfusate via probes (Terumo CDI™ 500 shunt sensor, Terumo, Leuven, Belgium) on the inflow and outflow tubing using an inline blood gas analyzer (CDI™ 500, Terumo, Borken, Germany). Oxygenation capacity was calculated using the formula PO2/FiO2. Temperature of the inflowing and outflowing perfusate was continuously measured, the latter considered to be equal to the graft temperature.
Finally, at the end of reperfusion, the perfused left lung and non-perfused right lung were weighed and evaporated overnight in an oven to a constant weight to measure wet–dry weight relationship. Extravascular lung water volumes (EVLW) were calculated as described by Pearce and co-workers [7] using the following equation: [(wet weight − dry weight)/wet weight] × 100%.
2.8 Statistics
Data analysis was performed using the software package Statistica 6.0. (StatSoft Inc., STATISTICA, www.statsoft.com).
All data are expressed as mean ± SEM. Friedman ANOVA was used to test for significant differences in time for MAP, HR, PAP and Plateau AwP in HBD and CONTROL during in situ assessment. Differences between both groups were analyzed with a Mann–Whitney U-test.
A Kruskall–Wallis ANOVA test with multiple comparisons was performed to look for differences in PVR, Plateau AwP, PO2/FiO2 and EVLW between HBD, CONTROL and NHBD during isolated reperfusion. p-values ≪0.05 were considered significant.
3 Results
3.1 Baseline conditions
Baseline values (HR, MAP, PAP and Plateau AwP) in all three groups were taken after completion of all surgical interventions 15 min before the start of the experimental protocol (time t = 0 min). Plateau AwP was recorded after a standard recruitment maneuver. No significant differences were observed between all groups (Table 1).
3.2 In situ evaluation in HBD and CONTROL
3.2.1 Hemodynamic changes
Brain death caused a hyperdynamic response in HBD compared with CONTROL. After intracranial pressure was increased in HBD, a transient significant rise in heart rate was observed from 15 until 60 min compared with baseline (95 ± 4 beats/min) with a peak at 15 min (175 ± 10 beats/min; p = 0.002). No time effect occurred within CONTROL. Heart rate was significantly different between HBD and CONTROL at 15, 30, 45 and 60 min (Fig. 2A ).
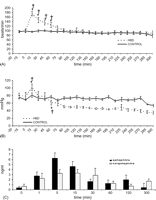
Panel A: heart rate in HBD and CONTROL during 5-h in situ observation interval following extradural balloon insertion (time t = 0 min). Data are presented as mean ± SEM. (*p ≪ 0.05 compared with baseline (time t = −15 min); †p ≪ 0.05 compared with CONTROL). Panel B: mean arterial pressure in HBD and CONTROL during 5-h in situ observation interval following extradural balloon insertion (time t = 0 min). Data are presented as mean ± SEM (*p ≪ 0.05 compared with baseline (time t = −15 min); †p ≪ 0.05 compared with CONTROL. Signs on the graph at time t = 60 min indicate the starting point from which values become significant for the remainder of the observation period). Panel C: plasma catecholamine concentrations in HBD during 5-h in situ observation interval following extradural balloon insertion (time t = 0 min). Data are presented as mean ± SEM.
Fifteen minutes after balloon inflation in HBD, MAP increased acutely to 102 ± 9 mmHg at 15 min compared with baseline (80 ± 7 mmHg; p = 0.01) and rapidly declined to 61 ± 7 mmHg at 60 min. A prolonged neurogenic hypotensive period was then observed in HBD with MAP significantly lower compared with CONTROL up to 5 h. MAP was stable within CONTROL throughout the entire interval (Fig. 2B).
3.2.2 Catecholamine concentrations
Increased levels of plasma catecholamines were documented following induction of brain death in HBD. Epinephrine and norepinephrine levels peaked significantly from baseline values by 1600% at 5 min and by 480% at 15 min, respectively (Fig. 2C). Baseline values in CONTROL did not differ significantly from HBD and remained stable throughout the entire interval (data not shown).
3.2.3 Pulmonary changes
In HBD, PAP fell significantly below baseline (21 ± 2 mmHg) starting at 150 min (16 ± 1 mmHg) until the end of the evaluation interval. Differences in PAP between HBD and CONTROL were not significant (Fig. 3A ).
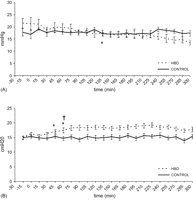
Panel A: pulmonary artery pressure in HBD and CONTROL during 5-h in situ observation interval following extradural balloon insertion (time t = 0 min). Data are presented as mean ± SEM (*p ≪ 0.05 compared with baseline (time t = −15 min). Sign on the graph at time t = 150 min indicates the starting point from which values become significant for the remainder of the observation period). Panel B: plateau airway pressure in HBD and CONTROL during 5-h in situ observation interval following extradural balloon insertion (time t = 0 min). Data are presented as mean ± SEM (*p ≪ 0.05 compared with baseline (time t = −15 min); †p ≪ 0.05 compared with CONTROL. Signs on the graph at time t = 60 min indicate the starting point from which values become significant for the remainder of the observation period).
A significant increase in Plateau AwP occurred in HBD starting at 45 min (17 ± 1 cmH2O) compared with baseline (15 ± 1 cmH2O). Plateau AwP was significantly higher in HBD compared with CONTROL starting at 60 min until the end of the observation period (Fig. 3B).
3.3 Ex vivo graft assessment in HBD, CONTROL and NHBD
3.3.1 Pulmonary vascular resistance
Pulmonary vascular resistance during controlled reperfusion with a continuous inflow pressure of 15 mmHg was significantly higher in HBD compared with CONTROL and NHBD starting at 40 min until the end of reperfusion. PVR in NHBD was lower compared with CONTROL without achieving the level of significance (Fig. 4 ).
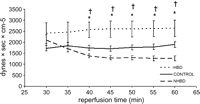
Pulmonary vascular resistance in HBD, CONTROL and NHBD from 30 until 60 min of isolated reperfusion (*p ≪ 0.05 in HBD compared with CONTROL; †p ≪ 0.05 in HBD compared with NHBD). There were no significant differences between NHBD and CONTROL.
3.3.2 Plateau airway pressure
During the whole evaluation period, Plateau AwP in HBD was significantly higher compared with CONTROL. Values in NHBD were lower compared with HBD and higher compared with CONTROL, but differences were not significant (Fig. 5 ).
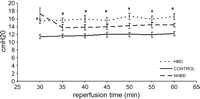
Plateau airway pressure in HBD, CONTROL and NHBD from 30 until 60 min of isolated reperfusion (*p ≪ 0.05 in HBD compared with CONTROL). Values in NHBD stabilized between HBD and CONTROL but were not significantly different.
3.3.3 Oxygenation capacity
There were no significant differences in PO2/FiO2 between HBD, CONTROL and NHBD during the evaluation period (Fig. 6 ).
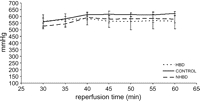
PO2/FiO2 in HBD, CONTROL and NHBD from 30 until 60 min of isolated reperfusion. No significant differences were observed.
3.3.4 Extravascular lung water
There were no significant differences in EVLW between HBD, CONTROL and NHBD before (=right lung) (Fig. 7A ) and after (=left lung) (Fig. 7B) ex vivo reperfusion.
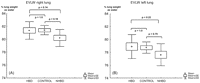
Extravascular lung water accumulation between HBD, CONTROL and NHBD. Panel A: right lung (before reperfusion). Panel B: left lung (after reperfusion).
4 Discussion
This study is the first that compared two types of lung donors. Porcine lung grafts from brain-dead and asystolic donors were compared with control animals. Graft function was assessed in an isolated reperfusion model looking at physiological variables. We demonstrated that brain death in HBD impairs graft function upon reperfusion more importantly compared with a limited warm ischemic interval in NHBD. This was reflected by an increased PVR and Plateau AwP in our model.
The first objective of this study was to develop a reproducible large animal model of explosive brain death to study donor lung injury. We used domestic pigs because of our extensive experience with isolated reperfusion and ventilation of porcine lungs [3,8,9]. Multiple small and large animal models have been described using different methods to induce brain death. Most of them did not study pulmonary function [10]. Explosive brain death in HBD leads to significant hemodynamic changes. The acute loss of brain stem function is immediately followed by a sympathetic or autonomic storm. This hyperdynamic phase is caused by an intense adrenergic activity.
In our model, intracranial pressure was acutely increased and sustained above systemic arterial pressure since this is the most relevant way to study the effects of brain death on donor organs.
Expansion of the supratentorial mass by balloon inflation led to brain stem herniation and global brain ischemia. This was confirmed by absent brain stem reflexes (corneal and pupillary light reflex).
We observed significant arterial hypertension with tachycardia as an essential part of the hyperdynamic state, rapidly followed by a cardiovascular collapse phase with sustained neurogenic hypotension. We, as well as others [11,12], did not observe an early episode of bradycardia, just prior to the hyperdynamic changes as was originally described by Cushing in 1902, and what is now referred to as the ‘cushing response’. The increase in hemodynamics in HBD during autonomic storm was associated with changes in circulating plasma catecholamine concentrations.
As we were interested in the early physiological events following brain death we did not include a protocol of volume expansion or vasoactive support to correct and stabilize these changes in blood pressure. This may have had an impact on our findings in comparison with the clinical situation.
Our second objective was to compare the quality of the pulmonary graft between HBD and NHBD during early reperfusion.
Our NHBD animals were sacrificed by myocardial fibrillation resulting in sudden cessation of organ perfusion. No attempt for cardio-pulmonary resuscitation was included in the protocol and animals were not heparinized, resembling a NHBD category I according to the Maastricht classification [2]. Grafts were left untouched inside the cadaver during 1-h post-mortem. This warm ischemic interval is well tolerated by the lung through maintenance of cellular respiration by diffusion of the oxygen reserve in the alveoli [13]. In situ preservation was prolonged by means of topical cooling for 4 h. Our group previously demonstrated that this technique can safely extend the ischemic interval by 6 h inside the cadaver [8,9]. This enables optimal organization of organ retrieval by the transplant center.
We admit that the animals in our NHBD group were exposed to only a short agonal phase. Mauney and co-workers [14] found that a pre-mortem hypoxic insult of 7.2 min following disconnection of ventilation did not cause additional injury. In the clinical scenario of a category III NHBD [2], however, longer hypoxic intervals prior to cardiac arrest are no exception and may indeed compromise graft quality. Pre-mortem hemodynamic instability resulting from massive hemorrhage or myocardial infarction may also impair graft function as was shown by Tremblay and co-workers [15] in a rat model. Hypoxia and ischemia of other organ systems such as the splanchnic circulation also results in an increase of inflammatory markers. However, our primary interest was to compare solely the insult caused by the warm ischemic period with the injury accompanying brain death in the HBD. The contribution of the agonal phase prior to circulatory arrest should be clarified in further experiments. This may differ according to the mode of death in NHBD (asphyxia, exsanguination, cardiogenic shock, sustained resuscitation).
Pulmonary graft function in all groups was evaluated in our previously described model of isolated reperfusion, validated to identify early graft injury [3,8,9]. We observed a significant increase in PVR in HBD compared with CONTROL and NHBD. It is likely that this increased vascular resistance will persist after transplantation and may contribute to the development of graft failure and right ventricular dysfunction. Plateau AwP was also significantly higher in HBD compared with CONTROL. Although not significant, Plateau AwP was better in NHBD compared with HBD. We could not show any differences in accumulation of extravascular lung water and oxygenation capacity between all groups in this study. We believe that this is related to the short reperfusion period (60 min) under controlled circumstances with a limited inflow pressure (15 mmHg) and a high oncotic activity of our reperfusion solution (8% human albumin). The controlled setting of our reperfusion model is necessary to limit additional injury during evaluation.
The predictive value of all parameters in our isolated reperfusion model will need further validation in a transplant model.
So far, most experimental studies looking at the detrimental effects of donor brain death have focused on heart, kidney and liver transplantation. With regard to the lung, injury in the donor can develop from mechanical ventilation, trauma, aspiration or infection but also owing to pathophysiological changes directly related to brain death itself. Most evidence about this relation is provided in studies on neurogenic pulmonary edema [16–21], a form of edema that is precipitated by a variety of injuries to the central nervous system such as subarachnoid hemorrhage, cerebral seizures and head trauma.
To explain the pulmonary events that occur in neurogenic pulmonary edema, many investigators have focused on the hyperdynamic phase shortly after brain death in association with elevated catecholamine levels [21]. The blood pools in the pulmonary vasculature as a result of increased systemic vascular resistance. This is associated with increased pulmonary arterial and left atrial pressures. The elevated capillary pressure is responsible for the development of hydrostatic edema [20]. Eventually, structural damage to the alveolar-capillary membrane increases permeability [18,19]. We also found an increased level of catecholamines after induction of brain death.
In contrast to these previous reports, our study did not show a significant increase in pulmonary artery pressure in HBD. This correlates with the findings of Hoff and co-workers [17] and Beckman and co-workers [16] that other mechanisms may be involved. In a series of experiments these authors questioned the role of systemic and pulmonary hyperdynamic responses as the only cause of neurogenic pulmonary edema. A direct increase in capillary permeability by adrenergic mechanisms was identified in the absence of hydrostatic induced damage.
Besides the clear hemodynamic changes occurring in brain-dead subjects, it is now widely demonstrated that systemic inflammatory pathways are also activated. Multiple cytokines (IL-1, IL-2, IL-6, TNF-α, IFN-γ) are upregulated in peripheral organs [22] and IL-8 is enhanced in BAL-fluid from HBD donors [23]. Similar inflammatory changes are described in patients with acute lung injury [6]. This supports the theory that pathological changes in brain-dead donor lungs may be related to the development of acute lung injury.
During in situ measurement of graft function in HBD, we observed a significant increase in Plateau AwP. This parameter provides the best clinically available estimate of alveolar pressure, an important determinant of alveolar distention. This increased alveolar pressure may result from alveolar instability and collapse of alveoli, a phenomenon that has also been observed during the development of acute lung injury and lung edema. The increased values of Plateau AwP are therefore likely to represent the development of lung injury at the level of the alveoli, either by hemodynamic or inflammatory changes. No HBD animals, however, developed clinically evident alveolar edema prior to harvest and no differences in EVLW were seen between HBD and CONTROL prior to reperfusion.
In a recent review [24] and further rat study [25], Avlonitis and co-workers focused on the interactions between adrenergic, hemodynamic and inflammatory pathways. Pro-inflammatory mediators may be directly released from the brain. Rapid changes in hemodynamics may induce endothelial shear stress with inflammatory upregulation and systemic hypoperfusion can trigger the release of cytokines. Hemodynamic correction of the hypertensive phase with α-adrenergic antagonists and of the hypotensive phase with noradrenaline attenuated inflammatory responses.
Finally, Ware and co-workers [26] recently described a high incidence of pulmonary arterial thrombosis and pulmonary infarction in lung grafts from brain-dead donors that were rejected for transplantation. This might also contribute to the increased PVR during isolated reperfusion in brain-dead animals. Of course, the occurrence of pulmonary thrombosis in warm ischemic grafts is also possible.
In further studies, we are now looking at alveolar liquid clearance to clarify potential mechanisms and to formulate future treatment strategies to counteract neurogenic pulmonary edema.
In conclusion, we developed a reproducible large animal model of explosive brain death to study donor lung injury. Pulmonary vascular resistance in lungs from HBD was worse compared with organs from NHBD in our model of isolated reperfusion. The other graft parameters did not reflect any worse impact of warm ischemia on the development of donor lung injury when compared with brain death. This study further supports the use of lungs from NHBD in pulmonary transplantation to increase the potential donor pool.
Presented at the joint 19th Annual Meeting of the European Association for Cardio-thoracic Surgery and the 13th Annual Meeting of the European Society of Thoracic Surgeons, Barcelona, Spain, September 25–28, 2005.
Winner of the 2005 Alessandro Ricchi Transplantation Services Award of The European Association for Cardio-thoracic Surgery.
Acknowledgments
This work was supported by grants from FWO-Vlaanderen (G.0576.06) and from Katholieke Universiteit Leuven (OT/03/55). Professor Dr D. Van Raemdonck is a Senior Clinical Investigator of the Fund for Scientific Research, Flanders, Belgium (G.3C04.99).
The authors would like to thank Professor Dr W. Flameng, Professor Dr P. Herijgers, Professor Dr J. Van Loon, Mevr M. Mathys, Mevr N. Jannis for their expert help, technical and administrative assistance. We thank Dr T. Nawroth for his advice in statistics.
Appendix A
Conference discussion
Dr M. Erasmus (Groningen, The Netherlands): You state that non-heart-beating donation had less injury, but your non-heart-beating donation was by ventricular fibrillation. We now use, in Holland, non-heart-beating donation after switch-off procedure, and then you see the same cascade of inflammation and hemodynamic instability as seen after brain death. Do you think that will change your outcome when you use an organ donor?
Dr Neyrinck: I fully agree with your comment. We know that in our model we studied the warm ischemic damage, but in the non-heart-beating donor, the agonal phase can contribute as well to the donor lung injury.
It was our primary end-point to study the warm ischemic damage by creating this optimal non-heart-beating donor.
Dr A. Pavie (Paris, France): If we follow your conclusion, owing to the lack of donor, it is a good idea to try to increase non-beating heart procurement.
Dr Neyrinck: Indeed, this study further supports the use of non-heart-beating donors in lung transplantation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}