





Abstract
Inflammatory bowel disease [IBD] presenting in early childhood is extremely rare. More recently, progress has been made to identify children with monogenic forms of IBD predominantly presenting very early in life. In this study, we describe the heterogeneous phenotypes and genotypes of patients with IBD presenting before the age of 2 years and establish phenotypic features associated with underlying monogenicity.
Phenotype data of 62 children with disease onset before the age of 2 years presenting over the past 20 years were reviewed. Children without previously established genetic diagnosis were prospectively recruited for next-generation sequencing.
In all, 62 patients [55% male] were identified. The median disease onset was 3 months of age (interquartile range [IQR]: 1 to 11). Conventional IBD classification only applied to 15 patients with Crohn’s disease [CD]-like [24%] and three with ulcerative colitis [UC]-like [5%] phenotype; 44 patients [71%] were diagnosed with otherwise unclassifiable IBD. Patients frequently required parenteral nutrition [40%], extensive immunosuppression [31%], haematopoietic stem-cell transplantation [29%], and abdominal surgery [19%]. In 31% of patients, underlying monogenic diseases were established [EPCAM, IL10, IL10RA, IL10RB, FOXP3, LRBA, SKIV2L, TTC37, TTC7A]. Phenotypic features significantly more prevalent in monogenic IBD were: consanguinity, disease onset before the 6th month of life, stunting, extensive intestinal disease and histological evidence of epithelial abnormalities.
IBD in children with disease onset before the age of 2 years is frequently unclassifiable into Crohn’s disease and ulcerative colitis, particularly treatment resistant, and can be indistinguishable from monogenic diseases with IBD-like phenotype.
1. Introduction
Very early-onset inflammatory bowel disease [VEOIBD] refers to children with IBD diagnosis established before the 6th year of life.1 This age group represents the minority of worldwide reported IBD cases, with an estimated incidence of 4.37 and a prevalence of 14 per 100000 children.2 Similar to adults with IBD, children present with abdominal pain, intestinal bleeding, diarrhoea, and weight loss.3 Of additional concern in young children, however, are the effects of chronic inflammation on growth and global development.4 In paediatric patients it is therefore paramount to establish effective diagnostic and management strategies early in the disease course,5 even more so as children are more likely to present with extensive and treatment-resistant disease compared with adults with IBD.6,7 IBD classification has recently been modified to address the younger age group, but this might not be sufficient to accommodate the heterogeneous phenotype of VEOIBD.8 IBD-like intestinal inflammation can also present as a feature of a phenotypically diverse group of monogenic conditions including primary immunodeficiencies affecting T and/or B cells, phagocyte defects, hyper- and autoinflammatory disorders, immune regulation, and epithelial barrier defects.9 The overall incidence and prevalence of monogenic IBD is still unknown, and the feasibility of comprehensive genetic screening in IBD has yet to be established.10 To our knowledge, this study to date reports the largest cohort of genetically screened patients with IBD onset before the age of 2 years.
2. Materials and Methods
Case notes and electronic case records and databases from 62 patients with disease onset before the age of 2 years, managed at our centre over the past 20 years, were reviewed. Anthropometric data, clinical features and laboratory results at presentation were extracted. After exclusion of infectious and allergic/eosinophilic gastrointestinal conditions, we classified the disease according to the clinical and histological phenotype established during the observation period [Table 1]. For the unclassified IBD cohort, ‘extensive disease’ [ED] was defined as evidence of acute and/or chronic intestinal inflammation of the upper [L4a, Paris equivalent] and lower gastrointestinal [GI] tract [L2/3, Paris equivalent]. The conventional definition for paediatric and adult IBDU [IBD restricted to the colon without features favouring CD- or UC-like disease] was expanded for our patient cohort to accommodate all unclassifiable inflammatory phenotypes similar to recently published reviews on early forms of IBD.5
Disease phenotypes and abbreviations.11
| Disease phenotype | Abbreviation | Definition |
|---|---|---|
| Infantile-onset IBD | IOIBD | Histological evidence of chronic [with/without acute] intestinal inflammation anywhere in the gastrointestinal [GI] tract and symptommonset before the age of 2 years |
| Crohn’s disease-like | CD-like | Endoscopic evidence of non- contiguous aphthous or linear ulcers in the GI tract, histological evidence of non-caeseating granulomas and perianal or fistulising disease |
| Ulcerative colitis-like | UC-like | Continuous mucosal inflammation of the colon, starting rectally without small bowel involvement other than backwash ileitis, and no CD-specific features |
| IOIBD unclassifiable | IOIBDU | IOIBD and absence of features favouring CD- or UC-like disease |
| Disease phenotype | Abbreviation | Definition |
|---|---|---|
| Infantile-onset IBD | IOIBD | Histological evidence of chronic [with/without acute] intestinal inflammation anywhere in the gastrointestinal [GI] tract and symptommonset before the age of 2 years |
| Crohn’s disease-like | CD-like | Endoscopic evidence of non- contiguous aphthous or linear ulcers in the GI tract, histological evidence of non-caeseating granulomas and perianal or fistulising disease |
| Ulcerative colitis-like | UC-like | Continuous mucosal inflammation of the colon, starting rectally without small bowel involvement other than backwash ileitis, and no CD-specific features |
| IOIBD unclassifiable | IOIBDU | IOIBD and absence of features favouring CD- or UC-like disease |
Disease phenotypes and abbreviations.11
| Disease phenotype | Abbreviation | Definition |
|---|---|---|
| Infantile-onset IBD | IOIBD | Histological evidence of chronic [with/without acute] intestinal inflammation anywhere in the gastrointestinal [GI] tract and symptommonset before the age of 2 years |
| Crohn’s disease-like | CD-like | Endoscopic evidence of non- contiguous aphthous or linear ulcers in the GI tract, histological evidence of non-caeseating granulomas and perianal or fistulising disease |
| Ulcerative colitis-like | UC-like | Continuous mucosal inflammation of the colon, starting rectally without small bowel involvement other than backwash ileitis, and no CD-specific features |
| IOIBD unclassifiable | IOIBDU | IOIBD and absence of features favouring CD- or UC-like disease |
| Disease phenotype | Abbreviation | Definition |
|---|---|---|
| Infantile-onset IBD | IOIBD | Histological evidence of chronic [with/without acute] intestinal inflammation anywhere in the gastrointestinal [GI] tract and symptommonset before the age of 2 years |
| Crohn’s disease-like | CD-like | Endoscopic evidence of non- contiguous aphthous or linear ulcers in the GI tract, histological evidence of non-caeseating granulomas and perianal or fistulising disease |
| Ulcerative colitis-like | UC-like | Continuous mucosal inflammation of the colon, starting rectally without small bowel involvement other than backwash ileitis, and no CD-specific features |
| IOIBD unclassifiable | IOIBDU | IOIBD and absence of features favouring CD- or UC-like disease |
Over a 3-year period, we prospectively recruited candidates for further molecular evaluation, including next-generation sequencing [NGS] as described previously.10 We thereby focused on already established monogenic IBD genes published elsewhere.5,9,10 Patients were informed and consented for functional studies and NGS as part of the PETIT Study [Patients with Early-onseT Intestinal inflammaTion]. Ethical approval was obtained from the National Research Ethics Service Committee London.
2.1. Statistical analysis
IBM SPSS Statistics for Windows, version 22 [Armonk, NY] was used to perform statistical analysis. Continuous data are presented as medians with interquartile ranges, and categorical data are presented as rates and proportions.
The Mann–Whitney U test and Kruskal–Wallis test were used to compare continuous data between groups. Spearman’s rho was used to interrogate correlation between two continuous data sets. To compare proportions between groups, we used Pearson chi-square and Fisher’s exact test where appropriate. All tests were two-tailed and significance level was set at 5%. Weights and heights were converted to weight-for-age, height-for-age and body mass index [BMI] z-scores using WHO Anthro- and WHO Anthro Plus software.
3. Results
Between 1995 and 2015, 62 patients with disease onset before the age of 2 years were treated at our centre, 55% of whom were male (see Figure 1). For 58% of children [36/62], diagnostic investigations [endoscopy and laboratory work-up] were performed at our centre, with the remainder being referred from other gastroenterology units from within the UK [n = 18], Central Europe [n = 4] and the Middle East/Asia [n = 4].
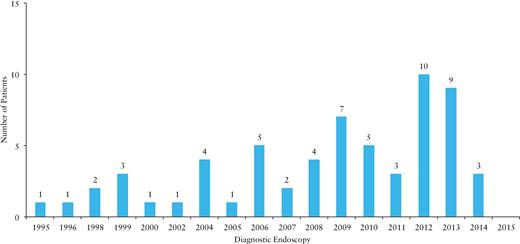
Year of first endoscopic evaluation of all 62 patients treated at our centre between 1995 and 2015.
Of our patients, 52% were White Europeans, 16% were Middle Eastern/Arab States, 8% Pakistani, 8% Indian, 6% Bangladeshi, 5% African, and 5% of mixed ethnic origin; 29% were offspring from consanguineous unions; 18% had a positive family history of IBD amongst first-degree relatives; and 15% had at least one sibling affected with IBD. The overall mortality rate was 5% (first patient: IPEX syndrome, idiopathic pneumonitis with multiorgan failure 12 months after haematopoietic stem cell transplantation [HSCT]; secondnd patient: IL10RA deficiency, pulmonary hypertension 6 weeks after HSCT; third patient: no causative mutation found on whole exome sequencing, fulminant pneumonia/sepsis and background of chronic lung disease).
The median observed disease duration for all 62 patients [diagnostic endoscopy to last contact] was 56 months [IQR: 29 to 107]. The median disease onset was at 3 months of age [IQR: 1 to 11]. Figure 2 describes the reported gastrointestinal symptoms at disease onset as well as weight-for-age [available for 60/62 patients], height-for-age [available for 57/62 patients], and BMI z-scores [available for 54/62 patients] from measurements taken within 4 weeks before diagnostic endoscopy. The median weight-for-age z-score was -1.11 [IQR: -2.65 to -0.06], the median height-for-age z-score was -1.83 [IQR: -2.94 to -0.47], and the median of BMI z-score was -0.5 [IQR: -1.40 to 0.85]; 30% of children had moderate or severe malnutrition, 48% had height-z-scores of less than -2 and 13% had BMI z-scores of less than -2. Children with earlier disease onset presented with lower weight-for-age [rho = 0.373, p = 0.003], height-for-age [rho = 0.295, p = 0.026], and BMI z-scores [rho = 0.312, p = 0.022].
![Symptoms reported at diagnosis for 62 patients with IOIBD. Light grey: non-monogenic cohort [n = 43], dark grey: monogenic cohort [n = 19]. Anthropometric data taken within 4 weeks before diagnostic endoscopy: graphs show height-for-age, weight-for-age and BMI z-scores for children diagnosed with CD-like, UC-like, and unclassifiable IOIBD [IOIBDU]. Circles represent non-monogenic cases, triangles represent monogenic cases. Dotted horizontal lines represent 50th, 9th, and 0.4th centiles. BMI, body mass index; CD, Crohn’s disease; UC, ulcerative colitis.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ecco-jcc/11/1/10.1093_ecco-jcc_jjw118/4/m_jjw11802.jpeg?Expires=1750704237&Signature=OaSZcA4LIKFjukRcqyfG76OOnKgmGJV-Fc8py1cdS1nDY7FBnyT30SYoxSBSIqXk55kJfUAkbZ2M452mb3a-sHnzhCG8RrHytAhg~g9BFUZGL6yNNyRvedVCW2FQ3~40GoxxpqYfYRdLa40JHi~Cg1P31N9FV-K3IMVMzEfdZDdgBnakomZv6PIYW7NvDuvUGUA4gmrm497J4X3gq9~FinoEOC5KgdWUSEYkb7pcZF27F1qQBa4lAqCyhV6dQCwo7jvXBmLCdqJF4~Gn8NO8mZ~8zHO2O9dDvUpadz6CeK~KC~5Dx1I1Sj44YRaMTc0XRDyf6H2bykEVHMXqlSpSPA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Symptoms reported at diagnosis for 62 patients with IOIBD. Light grey: non-monogenic cohort [n = 43], dark grey: monogenic cohort [n = 19]. Anthropometric data taken within 4 weeks before diagnostic endoscopy: graphs show height-for-age, weight-for-age and BMI z-scores for children diagnosed with CD-like, UC-like, and unclassifiable IOIBD [IOIBDU]. Circles represent non-monogenic cases, triangles represent monogenic cases. Dotted horizontal lines represent 50th, 9th, and 0.4th centiles. BMI, body mass index; CD, Crohn’s disease; UC, ulcerative colitis.
3.1. Endoscopy
Diagnostic endoscopy data were available for 57/62 patients. Children underwent diagnostic endoscopy at a median age of 13 months [IQR: 5 to 28].
Diagnostic delay [onset of symptoms to diagnostic endoscopy > 6 months] was seen in 44% [25/57] of patients. In this sub-group, suspected conditions before endoscopic assessment were: allergic GI disease [n = 12], chronic idiopathic constipation [n = 2], other multi-system diseases with intermittent non-bloody diarrhoea [n = 5], and intermittent non-bloody diarrhoea not further specified [n = 6]. Perianal disease [n = 9] was not observed in the diagnostic delay cohort, with a median time from onset of symptoms to diagnostic endoscopy of 3 months [IQR: 1 to 5].
For patients diagnosed externally [26/62], the first endoscopy performed at our centre was at a median age of 44 months [IQR: 14 to 67].
3.2. Histology
Longitudinal histology data for all patients were available for a median observational period of 16 months [IQR 0 to 45]. Patient data sets [clinical examination, cumulative endoscopy, and histology reports] were investigated for features suggestive of CD- or UC-like disease. Only 18 [29%] patients were classifiable as such according to the latest published international consensus.11 CD phenotypes were established according to the Paris Classification. All 15 patients with CD-like phenotype had upper GI involvement [L4a] and pancolonic disease among those 60% [9/15] involving the terminal ileum [L3]. One patient had a stricturing phenotype [B2], two had penetrating disease [B3], and one child had both [B2B3]. Perianal disease [p] was seen in 60% [9/15] of patients. All three children with UC-like phenotype had pancolonic disease [E4].
A total of 71% [44/62] of study participants had an unclassifiable disease pattern [IOIBDU]. In the IOIBDU group, extensive disease (ED, inflammation of the upper [L4a, Paris equivalent] and lower GI tract [L2/3, Paris equivalent]) was established in 77% [34/44]. Disease distribution and additional histological/clinical features are summarised in Table 2. Epithelial abnormalities were defined as morphologically abnormal intestinal epithelial layer [epithelial shedding, tufting] and or evidence of florid epithelial cell apoptosis. Monogenic diseases were established in 14 patients with IOIBDU and 93% [13/14] developed extensive disease [ED].
Cumulative clinical and histological features and anatomical distribution of inflammation in patients with unclassifiable IOIBD.
| Disease distribution IOIBDU | Epithelial abnormality | Stricturing disease | Villus blunting or atrophy | Monogenic IBD |
|---|---|---|---|---|
| PLOT [n = 7] | 0 | 3 | 3 | 1 |
| PC [n = 3] | 0 | 0 | 0 | 0 |
| ED [n = 34] TI involvement in 11/25 | 10 | 3 | 15 | 13 |
| Disease distribution IOIBDU | Epithelial abnormality | Stricturing disease | Villus blunting or atrophy | Monogenic IBD |
|---|---|---|---|---|
| PLOT [n = 7] | 0 | 3 | 3 | 1 |
| PC [n = 3] | 0 | 0 | 0 | 0 |
| ED [n = 34] TI involvement in 11/25 | 10 | 3 | 15 | 13 |
IOIBDU, infantile-onset inflammatory bowel disease unclassifiable; PLOT, proximal of the ligament of Treitz; PC, pancolonic; ED, extensive disease [PLOT and PC]; TI, terminal ileum; epithelial abnormality, epithelial shedding/tufting/abundant epithelial cell apoptosis.
Cumulative clinical and histological features and anatomical distribution of inflammation in patients with unclassifiable IOIBD.
| Disease distribution IOIBDU | Epithelial abnormality | Stricturing disease | Villus blunting or atrophy | Monogenic IBD |
|---|---|---|---|---|
| PLOT [n = 7] | 0 | 3 | 3 | 1 |
| PC [n = 3] | 0 | 0 | 0 | 0 |
| ED [n = 34] TI involvement in 11/25 | 10 | 3 | 15 | 13 |
| Disease distribution IOIBDU | Epithelial abnormality | Stricturing disease | Villus blunting or atrophy | Monogenic IBD |
|---|---|---|---|---|
| PLOT [n = 7] | 0 | 3 | 3 | 1 |
| PC [n = 3] | 0 | 0 | 0 | 0 |
| ED [n = 34] TI involvement in 11/25 | 10 | 3 | 15 | 13 |
IOIBDU, infantile-onset inflammatory bowel disease unclassifiable; PLOT, proximal of the ligament of Treitz; PC, pancolonic; ED, extensive disease [PLOT and PC]; TI, terminal ileum; epithelial abnormality, epithelial shedding/tufting/abundant epithelial cell apoptosis.
3.3. Laboratory findings
Laboratory markers at the time of first endoscopy were available for treatment-naïve patients diagnosed at our centre [36/62]: 61% [22/36] were anaemic, 31% [11/36] were hypoalbuminaemic, and 78% [28/36] had evidence of systemic inflammation established by raised C-reactive protein [CRP] [4/31], ESR [21/35] or platelet count [14/36]. Except for white cell counts found to be more frequently raised in the CD-like group, there was statistically no other significant difference between those parameters and the disease phenotypes [Figure S1, available as Supplementary data at ECCO-JCC online]. Faecal inflammatory markers were not consistently available owing to the retrospective study design and date of diagnosis in some children. Immunology work-up [lymphocyte subset, immunoglobulins and nitro blue tetrazolium test] revealed three children with IPEX disease presenting with high IgE levels [282 kU/l, 369 kU/l, and 34404 kU/l, normal range: 0–10 kU/l]. Three patients were found to have consistently abnormal immunology profiles: one patient with LRBA-deficiency presented with abnormally raised γδ-cells on T cell panel. Two TTC7A deficient patients presented with lower than normal T cell counts [reduced total CD3+ and CD4+ cells, respectively], in addition to low IgA levels in the former patient.
3.4. Genetics
Genetic screening in all 62 patients [Figure 3] revealed 19 cases [31%] of monogenic IBD [for detailed genetic data see Figure S2, available as Supplementary data at ECCO-JCC online]. Before the launch of the PETIT study, eight patients were diagnosed through Sanger sequencing [causative mutations established in four genes: IL10, IL10RA, IL10RB, FOXP3]. One patient with TTC7A deficiency was diagnosed externally by Sanger sequencing. Since the start of our study, 69% [37/54] of patients underwent TGPS and/or WES, with the remainder of 17 children screened through TGPS alone. Overall, NGS revealed 10 new diagnoses in six genes [EPCAM, IL10RA, TTC37, SKIV2L, LRBA, TTC7A]. Notably, in 37 patient samples screened by means of WES only two likely causative mutations were detected in novel genes [currently subject to functional follow-up studies]. Phenotypic variables were interrogated to establish the likelihood of underlying monogenic disease. Statistically significant clinical features for monogenic IBD are highlighted in Table 3.
![Age at disease-onset for all 19 monogenic IOIBD cases vs screening negative patients [n = 43]. IOIBD, infantile-onset inflammatory bowel disease.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ecco-jcc/11/1/10.1093_ecco-jcc_jjw118/4/m_jjw11803.jpeg?Expires=1750704237&Signature=Y7kVL~AzjfaFr6uGGVxjhLpxXjM8bmguO6lfbgdqxhGaTuAqSq5ffIxcW5WmML2IP1op5Hu7~7DtIEoPNA3BYnzlBQ2nKSB1wEegzpmdaj1mPJ6MhtWOWWVwETS281tQtpfbfQ90bET8hIl2j5dRSlpuGOx8hYvWyLyMT72Nd48ZnLhygdX0EMWTibXJpJFvQ3xY0~~IQiXi0KxOraRj1AqbJgsnTVn~rlggayXfmpQPporClpRgQ4YtmJfjbGVVH5EvoUs1y-rNuF85gsIOOX702dFVUWgXQEkeIkWwPILDXRmHPDCDWc~80S8ncYyQwOoMl0kz0cTAXcImP3sauQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Age at disease-onset for all 19 monogenic IOIBD cases vs screening negative patients [n = 43]. IOIBD, infantile-onset inflammatory bowel disease.
Proportion of cases with monogenic IBD compared within sub-cohorts defined by the presence/absence of specific features. Group 1: General background. Group 2: Features at presentation. Group 3: Cumulative clinical/histological features observed over median endoscopic follow-up time of 16 months. Group 4: Treatment deployed over median clinical follow-up time of 41 months.
| Group | Feature | Proportion of monogenic IBD within sub-cohorts defined by: | p-Value | |
|---|---|---|---|---|
| Feature present | Feature absent | |||
| 1 | Consanguinity | 61% | 18% | 0.002 |
| Male gender | 24% | 39% | 0.180 | |
| IBD family history [1st degree] | 55% | 25% | 0.058 | |
| 2 | Onset ≤ 6 months of age | 44% | 5% | 0.002 |
| Weight-for-age z-score < -3 [n = 60] | 42% | 25% | 0.252 | |
| Height-for-age z-score < -3 [n = 57] | 64% | 21% | 0.006 | |
| BMI z-score < -3 [n = 54] | 75% | 30% | 0.103 | |
| Blood per rectum | 24% | 36% | 0.315 | |
| Watery diarrhoea | 42% | 23% | 0.105 | |
| Anaemia [n = 36] | 18% | 21% | 1.000 | |
| Hypoalbuminaemia [n = 36] | 27% | 16% | 0.650 | |
| 3 | Extensive disease [ED] | 37% | 8% | 0.049 |
| Villus blunting/atrophy | 43% | 24% | 0.136 | |
| Granuloma | 0% | 35% | 0.093 | |
| Epithelial abnormality* | 90% | 19% | < 0.001 | |
| Perianal disease | 56% | 26% | 0.117 | |
| 4 | TIM+ | 26% | 33% | 0.623 |
| Abdominal surgery | 50% | 26% | 0.162 | |
| PN requirement | 60% | 11% | < 0.001 | |
| Group | Feature | Proportion of monogenic IBD within sub-cohorts defined by: | p-Value | |
|---|---|---|---|---|
| Feature present | Feature absent | |||
| 1 | Consanguinity | 61% | 18% | 0.002 |
| Male gender | 24% | 39% | 0.180 | |
| IBD family history [1st degree] | 55% | 25% | 0.058 | |
| 2 | Onset ≤ 6 months of age | 44% | 5% | 0.002 |
| Weight-for-age z-score < -3 [n = 60] | 42% | 25% | 0.252 | |
| Height-for-age z-score < -3 [n = 57] | 64% | 21% | 0.006 | |
| BMI z-score < -3 [n = 54] | 75% | 30% | 0.103 | |
| Blood per rectum | 24% | 36% | 0.315 | |
| Watery diarrhoea | 42% | 23% | 0.105 | |
| Anaemia [n = 36] | 18% | 21% | 1.000 | |
| Hypoalbuminaemia [n = 36] | 27% | 16% | 0.650 | |
| 3 | Extensive disease [ED] | 37% | 8% | 0.049 |
| Villus blunting/atrophy | 43% | 24% | 0.136 | |
| Granuloma | 0% | 35% | 0.093 | |
| Epithelial abnormality* | 90% | 19% | < 0.001 | |
| Perianal disease | 56% | 26% | 0.117 | |
| 4 | TIM+ | 26% | 33% | 0.623 |
| Abdominal surgery | 50% | 26% | 0.162 | |
| PN requirement | 60% | 11% | < 0.001 | |
IBD, inflammatory bowel disease; BMI, body mass index; PN, parenteral nutrition; TIM+, tumour necrosis factor alpha [TNFα]-blockade and first-line immunomodulator + second-line immunomodulator. In bold: p-values of <0.05.
*For definition of abnormal epithelial morphology, see above.
Proportion of cases with monogenic IBD compared within sub-cohorts defined by the presence/absence of specific features. Group 1: General background. Group 2: Features at presentation. Group 3: Cumulative clinical/histological features observed over median endoscopic follow-up time of 16 months. Group 4: Treatment deployed over median clinical follow-up time of 41 months.
| Group | Feature | Proportion of monogenic IBD within sub-cohorts defined by: | p-Value | |
|---|---|---|---|---|
| Feature present | Feature absent | |||
| 1 | Consanguinity | 61% | 18% | 0.002 |
| Male gender | 24% | 39% | 0.180 | |
| IBD family history [1st degree] | 55% | 25% | 0.058 | |
| 2 | Onset ≤ 6 months of age | 44% | 5% | 0.002 |
| Weight-for-age z-score < -3 [n = 60] | 42% | 25% | 0.252 | |
| Height-for-age z-score < -3 [n = 57] | 64% | 21% | 0.006 | |
| BMI z-score < -3 [n = 54] | 75% | 30% | 0.103 | |
| Blood per rectum | 24% | 36% | 0.315 | |
| Watery diarrhoea | 42% | 23% | 0.105 | |
| Anaemia [n = 36] | 18% | 21% | 1.000 | |
| Hypoalbuminaemia [n = 36] | 27% | 16% | 0.650 | |
| 3 | Extensive disease [ED] | 37% | 8% | 0.049 |
| Villus blunting/atrophy | 43% | 24% | 0.136 | |
| Granuloma | 0% | 35% | 0.093 | |
| Epithelial abnormality* | 90% | 19% | < 0.001 | |
| Perianal disease | 56% | 26% | 0.117 | |
| 4 | TIM+ | 26% | 33% | 0.623 |
| Abdominal surgery | 50% | 26% | 0.162 | |
| PN requirement | 60% | 11% | < 0.001 | |
| Group | Feature | Proportion of monogenic IBD within sub-cohorts defined by: | p-Value | |
|---|---|---|---|---|
| Feature present | Feature absent | |||
| 1 | Consanguinity | 61% | 18% | 0.002 |
| Male gender | 24% | 39% | 0.180 | |
| IBD family history [1st degree] | 55% | 25% | 0.058 | |
| 2 | Onset ≤ 6 months of age | 44% | 5% | 0.002 |
| Weight-for-age z-score < -3 [n = 60] | 42% | 25% | 0.252 | |
| Height-for-age z-score < -3 [n = 57] | 64% | 21% | 0.006 | |
| BMI z-score < -3 [n = 54] | 75% | 30% | 0.103 | |
| Blood per rectum | 24% | 36% | 0.315 | |
| Watery diarrhoea | 42% | 23% | 0.105 | |
| Anaemia [n = 36] | 18% | 21% | 1.000 | |
| Hypoalbuminaemia [n = 36] | 27% | 16% | 0.650 | |
| 3 | Extensive disease [ED] | 37% | 8% | 0.049 |
| Villus blunting/atrophy | 43% | 24% | 0.136 | |
| Granuloma | 0% | 35% | 0.093 | |
| Epithelial abnormality* | 90% | 19% | < 0.001 | |
| Perianal disease | 56% | 26% | 0.117 | |
| 4 | TIM+ | 26% | 33% | 0.623 |
| Abdominal surgery | 50% | 26% | 0.162 | |
| PN requirement | 60% | 11% | < 0.001 | |
IBD, inflammatory bowel disease; BMI, body mass index; PN, parenteral nutrition; TIM+, tumour necrosis factor alpha [TNFα]-blockade and first-line immunomodulator + second-line immunomodulator. In bold: p-values of <0.05.
*For definition of abnormal epithelial morphology, see above.
3.5. Treatment
Medications used to treat IOIBD included corticosteroids [92%], immunomodulators (84%; azathioprine, 6-mercaptopurine [6-MP], methotrexate, cyclosporine, tacrolimus, sirolimus, thalidomide), aminosalicylates [58%], and biologicals [39%; infliximab, adalimumab]; 31% of patients [19/62] received more than combination therapy of one first-line immunosuppressant drug [in 88% of cases: azathioprine/6-mercaptopurine] and tumour necrosis factor alpha [TNFα] blockade [infliximab or adalimumab] within the median time of observed disease duration of 56 months [TNFα-blockade and first-line immunomodulator + additional immunomodulator = TIM+] [Figure 4].
![Summary of patient characteristics, therapeutics, and outcome measures. N/a, not available; Not appl., not applicable; DoD, [observed] duration of disease; PT, phenotype; ED, extensive disease; ST, steroids; AZA, zathioprine; 6-MP, 6 mercaptopurine; MTX, methotrexate; IFX, infliximab; ADA, adalimumab; CsA, cyclosporine A; Siro, sirolimus; Tac, tacrolimus; Thal, thalidomide; TIM+, tumour necrosis factor alpha [TNFα] blockade and first-line immunosuppressant + second-line immunosuppressant; PN, parenteral nutrition; HSCT, hstem cell transplantation; S, stoma; H/C, hemi-/colectomy; IR, intestinal resection; PEG, percutaneous endoscopic gastrostomy; PGA, physician global assessment [1 = inactive disease; 2 = mild disease; 3 = moderate disease; 4 = severe disease].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ecco-jcc/11/1/10.1093_ecco-jcc_jjw118/4/m_jjw11804.jpeg?Expires=1750704237&Signature=S3qHjSMU409-AbnEPiJgdbFU5jC4IaGJdIzNB2qcexgedHiddPIns6dMtLiLv4aNBaPoeO3i2ZlvCiz-Wh9Ek7uLD~N63jZYNSdqdjZOGbFNvAcmFflFv7Sv6bxCgtjOo4mkj-p0vl9RrVF2HuJSnOjK3R-81HP6Lvifsv~059DUKWv7sPj1uEjySIbcHf2Cm1lQOyyn49F-glHqRDmkuDDgpCTmxo7K6lIRYEuLPqYvwEIT0~6AUDUl01d-48uxtMMwfJ833bwMsCYWXWxT7sQIGUcPVCqE3gdMLDsoW7P4rlNOqkMty5ULu8iC1B-tUhlpHGm33VuouUnmj9zsYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Summary of patient characteristics, therapeutics, and outcome measures. N/a, not available; Not appl., not applicable; DoD, [observed] duration of disease; PT, phenotype; ED, extensive disease; ST, steroids; AZA, zathioprine; 6-MP, 6 mercaptopurine; MTX, methotrexate; IFX, infliximab; ADA, adalimumab; CsA, cyclosporine A; Siro, sirolimus; Tac, tacrolimus; Thal, thalidomide; TIM+, tumour necrosis factor alpha [TNFα] blockade and first-line immunosuppressant + second-line immunosuppressant; PN, parenteral nutrition; HSCT, hstem cell transplantation; S, stoma; H/C, hemi-/colectomy; IR, intestinal resection; PEG, percutaneous endoscopic gastrostomy; PGA, physician global assessment [1 = inactive disease; 2 = mild disease; 3 = moderate disease; 4 = severe disease].
*Diagnostic endoscopy not performed at our centre; **previously reported in the literature10,25,47,51; [x], possible monogenic inflammatory bowel disease [IBD] [functional work-up in progress].
3.6. Treatment outcome
Figures 4 and 5 provide summaries of all treatments applied from diagnosis until the end of the observational period. Comparable outcome measures were requirement for: parenteral nutrition, abdominal surgery, treatment escalation to TNFα blockade, and HSCT. Overall, 40% of children required parenteral nutrition [PN] throughout their monitored disease course [≥ 50% of calorie requirement for ≥ 28 days; introduced for enteral food intolerance/gut rest and/or inadequate weight gain]; 19% [12/62] of patients underwent abdominal surgery: hemi/subtotal colectomy with stoma formation [n = 9], stoma formation alone [n = 2], surgery for isolated small bowel atresias [n = 1]. Percutaneous feeding devices were used in 23% of children. HSCT was performed in 18 children of whom 13 underwent transplantation for failure to respond to conventional treatment and six patients for known HSCT-amenable genetic diagnosis [IPEX syndrome and LRBA-IL10/RA deficiency].
![Comparison of the use of different treatment modalities between children with CD-like phenotype and unclassified IOIBD [UC-like disease not included due to the statistically insignificant number of cases [n = 3]. At the time of writing, subtotal colectomy was performed in one patient with UC-like phenotype, one patient with UC-like disease required PN, and a separate child with UC required intensive immunosuppression [TIM+]. Light grey: non-monogenic cases, dark grey: monogenic cases. IOIBD, infantile-onset inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ecco-jcc/11/1/10.1093_ecco-jcc_jjw118/4/m_jjw11805.jpeg?Expires=1750704237&Signature=dIWYpfAVTt-Gdt~gsIA-0dZx9uUbzHkDJ~W6URmVmh3jrrcX~0hq~fXKVanEuz5aNiLU9xMT6BpFf6E2wQBaMLPKMupjjYl7GsGT9l1v~6yxEdENJXRPrwIhG552TeB6Ga0W0eYyn89Z1DlwK0HD5N33dna8JB1J2K6XgA8a~hxNP6msK8h1KnTtr0ZoqGGGf6sfuuevu2Eusnx5IC-Tn5U9PgZrYtq-dk4f1NYimkSZ5fp~egAhgPnWg3nCPs2oghjbGpplZUThXRMwkJqrpokbMkejUk7EqV6wCcNmKlkBYvimabaEO3DPZgI5UOs4AOPzT-2kUO~FV~ms8dEnEA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Comparison of the use of different treatment modalities between children with CD-like phenotype and unclassified IOIBD [UC-like disease not included due to the statistically insignificant number of cases [n = 3]. At the time of writing, subtotal colectomy was performed in one patient with UC-like phenotype, one patient with UC-like disease required PN, and a separate child with UC required intensive immunosuppression [TIM+]. Light grey: non-monogenic cases, dark grey: monogenic cases. IOIBD, infantile-onset inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis.
3.7. Extensive vs limited IOIBD
Children with extensive disease [ED] were more likely to require intensified immunosuppression [TIM+] [37% vs 8%, p = 0.049] when compared with the 13 cases with limited disease.
3.8. IOIBD requiring only first-line therapy vs need for treatment escalation
A total of 26% of children [16/62] did not require PN, abdominal surgery, or treatment escalation beyond immunomodulator monotherapy [4/9 CD, 1/3 UC, and 11/30 IOIBDU]. There was no statistically significant difference with regars to age at onset of disease (8 months [IQR: 3 to 15] vs 6 months [IQR: 1 to 13], p = 0.158) but weight-for-age z-scores at presentation were significantly higher when compared with children requiring treatment escalation [-0.80 vs -1.46, p = 0.033].
3.9. HSC transplanted vs non-transplanted IOIBD
Patients who underwent HSCT had earlier disease onset (2 months [IQR: 0 to 4] vs 6 months [IQR: 1 to 15], p = 0.030) and required more intensive immunosuppression [TIM+] when compared with the cohort of non-transplanted children [56% vs 20%, p = 0.007].
3.10. Monogenic vs non-monogenic IBD
Children with monogenic IBD more frequently required parenteral nutrition [79% vs 23%, p < 0.001] and HSCT [63% vs 14%, p < 0.001] when compared with the non-monogenic IBD group.
4. Discussion
Our study confirms that children with IOIBD present with clinical and laboratory features reminiscent of conventional IBD. However, these children have more disseminated intestinal inflammation, higher rates of treatment resistance, and rapid disease progression when compared with adolescent or adult onset IBD.7,12 Extensive disease has profound effects on growth and weight, which was confirmed by the results of our study: 30% of children presented with moderate or severe malnutrition and 48% of children were stunted at the time of diagnosis. Stunting however is not solely explained by malnutrition, given that various monogenic diseases can present with growth failure as part of a syndromic phenotype13,14 In addition we observed a high prevalence of anaemia which partly reflects the presence of micronutrient deficiencies, as highlighted in previous reports.15,16
4.1. IOIBD diagnosis and classification
In our study, the median time interval between onset of symptoms and diagnostic endoscopy was 6 months. Suspected allergic GI disease was the single most common reason for delayed endoscopic evaluation in our cohort. Allergic GI disease and IOIBD share common clinical features.17 A diagnosis of IOIBD should be considered in the absence of response to dietary modification and profound impact of the disease on weight and growth, as well as in the presence of other IBD-associated features such as perianal involvement. Blood laboratory markers cannot reliably differentiate either disease. More recently, faecal calprotectin has been established as a useful tool in paediatric IBD.18,19 A potential role in allergic GI disease has been highlighted, but more evidence is required to establish its role in distinguishing between individual inflammatory disorders.20
The evidence suggests that the prevalence of unclassifiable IBD phenotypes in children with early onset IBD is higher when compared with the older paediatric and adult IBD population.6,21,22 In 2014, the European Society of Paediatric Gastroenterology, Hepatology and Nutrition [ESPGHAN] published a widely adopted approach to diagnose suspected paediatric IBD. The IBDU phenotype is reserved for patients with exclusively colonic inflammation in the absence of features suggesting either UC or CD.11 Patients with extensive disease might therefore be labelled as CD even in the absence of typical CD features. Labelling IOIBD early in the disease course as CD or UC might also prevent the physician from considering further investigations. This is particularly worrying in the case of CD-like IOIBD, given that typical microscopic and macroscopic features such as perianal/fistulising disease, linear or serpentine ulceration, and granulomas have also been reported in a variety of monogenic conditions such as chronic granulomatous disease [CGD], IPEX syndrome, XIAP-, or IL10/R deficiency.23–26 Low prevalence and phenotypic heterogeneity of IOIBD, in particular in patients with unclassifiable IOIBD, poses a challenge with regard to both developing diagnostic and clinical guidance and establishing IOIBD-specific outcome predictors.
It has therefore been suggested that second-line investigations, such as immune work-up and genetic diagnostics, constitute important stratification tools for patients with IOIBD given that many monogenic diseases can present with an IBD-like phenotype.5,9
4.2. Immune work-up
There is a strong argument to investigate basic immune components and function, given that primary immune deficiencies can present with IBD-like disease.5,9 In our cohort, immunophenotyping guided the diagnosis in patients with LRBA-, FOXP3-, and TTC7A deficiency. LRBA-deficiency has been previously reported to present with IBD-like disease, autoimmunity, and an IPEX-like phenotype.27–29 All three patients with FOXP3 mutations presented with significantly elevated total IgE, consistent with previous reports on children with IPEX syndrome.30–34 IBD-like intestinal inflammation and associated immunodeficiency have been described in patients with TTC7A mutations.35,36 Other conditions, such as IL10 or IL10R deficiency, were not picked up by basic immune screening and required specific functional analysis.5,25–27
4.3. Genetic screening
Given the large number of potential candidate genes and overlapping phenotypes, single gene sequencing appears less and less appropriate in children with IOIBD. Costs, availability, and processing time of NGS have significantly improved over recent years, making it the screening modality of choice. In a recent publication we have compared the two different NGS platforms for genetic screening applied in this study.10 In brief, WES is suitable for novel gene discovery but provides less reliable gene coverage in the diagnostic setting when compared with TGPS. Whether expanding diagnostic genetic screening beyond the currently established IBD genes has an immediate impact on patient care remains to be established. In our cohort of genome-wide screened patients, only two harboured potentially promising novel candidate genes [currently undergoing functional assessment]. In our group of patients, a genetic diagnosis could be established in 31% of patients. As an international referral centre, our cohort might over-represent ethnic populations with higher prevalence of consanguinity. An increased pre-test probability will therefore overestimate the prevalence of monogenic IBD as compared with population-based studies. However, our experience and previously published data suggest that comprehensive genetic screening regardless of consanguinity or ethnicity can reveal unexpected genotypes10 and other variables as highlighted in this study, such as onset of disease under 6 months of age, stunting [height-for-age z-score < 3], extensive disease, and epithelial abnormalities, were significantly more prevalent in the monogenic IOIBD cohort.
Histological assessment revealed a distinct IOIBDU subgroup of children with abnormal epithelial morphology and concomitant chronic or acute on chronic mucosal inflammatory cell infiltration. This feature carried a high positive predictive value and should prompt the physician to consider screening for genes involved in epithelial integrity. Very early-onset intestinal inflammation can be due to the absence of EPCAM, which results in tufting enteropathy [TE].38,39 The epithelial changes in TE can initially be localised and subtle, with inflammatory cell infiltration being a dominant feature; hence the decision to include the EPCAM gene in our monogenic IBD screening.10,40 Mutations in the gene TTC7A have recently been reported to cause IBD in children, with stricturing intestinal disease and severe epithelial abnormalities on histology.35,36,41,42 Abnormal epithelial morphology [epithelial shedding/sloughing] and increased epithelial cell apoptosis were evident in all three of our patients. Prominent epithelial surface irregularity and increased apoptotic activity were also present in patients with SKIV2L and TTC37 mutations. Causative variants in TTC37 and SKIV2L genes, both involved in the RNA decay pathway of the cell [exosome complex],43 have previously been reported to cause tricho-hepato-enteric syndrome [THES].13–14
4.4. Treatments and outcome
Phenotypes and treatments used during the observation period of 56 months were reviewed. Notably, 26% of children with IOIBD did not require surgery, PN, or treatment escalation beyond first-line immunomodulator therapy. No statistical difference was established in observed disease duration and age at onset between this sub-group and the remaining 74% of patients. Comparing the presence of clinical features at diagnosis between both groups revealed a significantly higher weight-for-age z-score in the former group, suggesting that the nutritional status at diagnosis might be an indicative factor for long-term disease progression. Overall, our study revealed a high proportion of children needing extensive immunomodulation and PN, consistent with previous reports.44–46 HSCT was performed for failure to respond to conventional treatment or in children with previously known HSCT-amenable genetic diagnoses.34,47–49 Despite small case numbers, our results suggest that establishing a genetic diagnosis early in the disease course supports decision making and can reduce the need for abdominal surgery and the burden associated with prolonged immunosuppression in some children.48 The 5-year cumulative risk for bowel surgery for paediatric CD is approximately 20%.50 In our cohort, 40% of children with CD-like disease required bowel surgery. Among five children with IL10/R deficiency, three had extensive abdominal surgery and two were diagnosed within months after presentation and underwent HSCT before needing intensification of medical or surgical management.47–51 All three patients with TTC7A deficiency required bowel resections and stoma formation, as previously reported in the literature.35–36 In this group of children, preliminary data suggest that HSCT corrects the associated primary immune deficiency but does not significantly alter the intestinal disease progression [unpublished data].
Our study has limitations commonly associated with a retrospective study design. First, faecal inflammatory markers were not readily available throughout the majority of the period of observation. Second, evaluation of the extent of disease was established through endoscopy and histology reports, as small bowel imaging [MRE] was considered not feasible in this age group.
In conclusion, our data provide evidence that conventional IBD classification tools insufficiently address the heterogeneous phenotype of IOIBD. We therefore suggest applying descriptive disease terminology and having a low threshold for revision of conventional disease labels. Particularly children with symptom onset before the age of 2 years should undergo comprehensive immunological and genetic screening and a molecular diagnosis should be considered. Establishing a genetic diagnosis has obvious advantages and potentially enables the physician to tailor treatment, predict outcome, and counsel patient and parents effectively.34,47,52
Funding
This study was performed in partnership with GOSgene based at the UCL Institute of Child Health, and is supported by the National Institute for Health Research Biomedical Research Centre [NIHR BRC]. This report is independent research by the NIHR BRC Funding Scheme. The views expressed in this publication are those of the author[s] and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health. H. H. Uhlig is supported by the Crohn’s and Colitis Foundation of America, and the Leona M. and Harry B. Helmsley Charitable Trust.
Conflict of Interest
HHU has project collaborations with UCB Pharma, Eli Lilly; has received payment for lectures from Actelion, MSD, and Mitsubishi Tanabe Pharma Corporation; and has patents through EUROIMMUN and receives royalties from EUROIMMUN. JK, RD, MP, SD, HG, KR, SC, BH, SS, CJ, NA, EC, MG, GNJ, FK, ME, PLB, NJS, KG, CB and NS declare no conflict of interest related to this article.
Author Contributions
JK, CJ, and CB were responsible for analysing the WES data, SD performed and analysed the TGPS data. RD performed statistical analysis. RD, HG, KR, SC, BH, SS, NA, EC, MG, GNJ, and JK extracted patient data. Histological data were evaluated by NJS and JK. Immunological data were analysed by KG. HHU, PLB, FK, ME, and NS advised on the design of the study. JK drafted the first version of the manuscript, with subsequent critical appraisal from all listed authors.
Supplementary Data
Supplementary data are available at ECCO-JCC online.
Acknowledgments
We thank all children and their parents for participating in the PETIT Study. We also like to thank the Clinical Genetics and Inflammatory Bowel Disease teams at Great Ormond Street Children’s Hospital for their support in recruitment and sample management.
References
Author notes
Corresponding author: J. Kammermeier, MRCPCH, MD, University College London [Institute of Child Health] and Great Ormond Street Hospital, 30 Guilford Street, London WC1N 1EH, UK Email: [email protected]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}