






Abstract
The recombinant protein-based vaccine, NVX-CoV2373, demonstrated 89.7% efficacy against coronavirus disease 2019 (COVID-19) in a phase 3, randomized, observer-blinded, placebo-controlled trial in the United Kingdom. The protocol was amended to include a blinded crossover. Data to the end of the placebo-controlled phase are reported.
Adults aged 18–84 years received 2 doses of NVX-CoV2373 or placebo (1:1) and were monitored for virologically confirmed mild, moderate, or severe COVID-19 (onset from 7 days after second vaccination). Participants who developed immunoglobulin G (IgG) against nucleocapsid protein but did not show symptomatic COVID-19 were considered asymptomatic. Secondary outcomes included anti-spike (S) IgG responses, wild-type virus neutralization, and T-cell responses.
Of 15 185 participants, 13 989 remained in the per-protocol efficacy population (6989 NVX-CoV2373, 7000 placebo). At a maximum of 7.5 months (median, 4.5) postvaccination, there were 24 cases of COVID-19 among NVX-CoV2373 recipients and 134 cases among placebo recipients, a vaccine efficacy of 82.7% (95% confidence interval [CI], 73.3%–88.8%). Vaccine efficacy was 100% (95% CI, 17.9%–100.0%) against severe disease and 76.3% (95% CI, 57.4%–86.8%) against asymptomatic disease. High anti-S and neutralization responses to vaccination were evident, together with S-protein–specific induction of interferon-γ secretion in peripheral blood T cells. Incidence of serious adverse events and adverse events of special interest were similar between groups.
A 2-dose regimen of NVX-CoV2373 conferred a high level of ongoing protection against asymptomatic, symptomatic, and severe COVID-19 through >6 months postvaccination. A gradual decrease of protection suggests that a booster may be indicated.
EudraCT, 2020-004123-16.
The coronavirus disease 2019 (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in significant morbidity and mortality worldwide, with 608 million cases and 6.5 million deaths reported as of 16 September 2022 [1].
Vaccination remains a key element for pandemic control. International efforts have led to development of safe and effective COVID-19 vaccines that target the virus spike (S) glycoprotein, with 38 vaccine candidates currently in clinical use [2].
Efforts to control the COVID-19 pandemic have been hindered by emergence of several SARS-CoV-2 genotypic variants, including Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529). These viral strains have shown increased transmissibility, severity of clinical disease, and potential immunologic escape from COVID-19 vaccine protection [3–7]. The Alpha variant was the most prevalent strain in the United Kingdom between January 2021 and May 2021 but was gradually replaced by the Delta variant, reaching 90% of sequenced cases by June 2021 [8]. Delta was rapidly replaced by the Omicron variant starting in November 2021 [9, 10]. In this context of emerging variants, it is crucial to closely monitor longer-term vaccine efficacy.
Effectiveness of COVID-19 vaccines in preventing asymptomatic infection is also important when considering the overall impact of vaccine programs. Prevention of both symptomatic and asymptomatic infection is likely to have a larger impact on interruption of transmission than prevention of symptomatic disease alone. Currently, limited data from randomized trials are available on vaccine efficacy against asymptomatic disease; 63% efficacy was reported for the mRNA-1273 vaccine (compared with 93.2% against symptomatic illness) [11], 28.9% for the Ad26.COV2.S vaccine [12], and 22.2% to 49.3% (depending on vaccine dose and schedule) for the ChAdOx1 nCoV-19 vaccine [13].
NVX-CoV2373 is a recombinant, nanoparticle, protein-based vaccine with Matrix-M™ adjuvant. Two 5-μg doses of vaccine, administered 21 days apart, have demonstrated safety and immunogenicity in phase 1 and 2 trials [14, 15] and high efficacy in 2 phase 3 trials [16, 17]. The 2019nCoV-302 study is a phase 3, randomized, observer-blinded, placebo-controlled trial to evaluate efficacy, immunogenicity, and safety of the NVX-CoV2373 vaccine in preventing COVID-19 in adults aged 18–84 years in the United Kingdom. We previously reported 89.7% protection against all symptomatic SARS-CoV-2 infection in the primary event-driven analysis, including high efficacy against the Alpha variant [18] and 96.4% efficacy against non-Alpha strains. The trial included a planned blinded crossover, ending the placebo-controlled portion of the study (conducted from 29 March 2021 to 14 June 2021). Here, we provide study results for safety and efficacy through the end of the placebo-controlled period and previously unreported immunogenicity analyses.
METHODS
Trial Design and Participants
The methodology and full protocol for this trial have been previously published [16]. Briefly, we assessed the safety and efficacy of two 5-µg doses of NVX-CoV2373 or placebo, administered intramuscularly 21 days apart. This phase 3, randomized, observer-blinded, placebo-controlled trial was conducted at 33 sites across the United Kingdom. Eligible participants were men and nonpregnant women aged 18 to 84 years (inclusive) who were healthy or had stable chronic medical conditions including, but not limited to, human immunodeficiency virus (receiving effective antiretroviral therapy) and cardiac and respiratory diseases. Participants were randomly (1:1) assigned via block randomization to receive 2 doses of NVX-CoV2373 or placebo (normal saline) using a centralized interactive response technology system according to pregenerated randomization schedules. Randomization was stratified by site and by age ≥ 65 years. Key exclusion criteria included history of documented COVID-19 and treatment with immunosuppressive therapy.
Results from the planned primary event-driven analysis, which included a median of approximately 3 months of follow-up (data cutoff date, 29 January 2021), have been published [16]. The protocol was amended on 25 February 2021 to include a blinded crossover phase in which subsequent doses of study vaccine were administered from 29 March 2021 to 14 June 2021 so that all participants could receive active vaccine during the study. Participants could request to be unblinded at any time during the study, whereby they could choose to receive an authorized COVID-19 vaccine through the UK National Health Service, remain in the study for safety follow-up, or withdraw from the study entirely. Those who remained blinded entered the blinded crossover or chose to remain blinded and not enter the blinded crossover (Figure 1). For this analysis, safety and efficacy data from this ongoing phase 3 trial were assessed at a maximum of 7.5 months (median, 4.5) after study start.
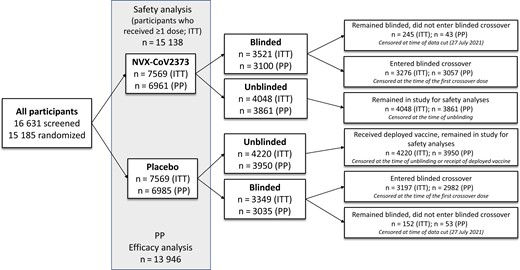
Participant disposition and status at the time of current analysis. Participants were randomly assigned in a 1:1 ratio to receive NVX-CoV2373 or placebo. Participants were able to request to be unblinded or to withdraw from the study at any time. Those who were unblinded in the placebo arm may have received a currently authorized vaccine from the National Health Service, while those who were unblinded in the NVX-CoV2373 arm may have chosen to remain in the study for follow-up. Those who remained blinded in either of the 2 arms entered the blinded crossover or chose to remain blinded in the study and not enter the blinded crossover. For the current analysis, participants were censored at the earliest of the date of unblinding (for any reason), date of receipt of an authorized coronavirus disease 2019 vaccine, date of entering the blinded crossover (date of receiving a third study dose), date of early withdrawal, date of death, or date of data cutoff (27 July 2021). Abbreviations: ITT, intention to treat; PP, per-protocol.
The trial protocol was approved by the North West–Greater Manchester Central Research Ethics Committee and was performed in accordance with the International Council for Harmonization Good Clinical Practice guidelines. Safety oversight was performed by an independent safety monitoring committee.
Safety
Safety data are reported for all participants who received at least 1 dose of vaccine or placebo. This includes serious adverse events (SAEs), AEs of special interest (AESIs), and related medically attended AEs (MAAEs) through the end of the placebo-controlled period (Supplementary Tables 1 and 2).
Efficacy
Efficacy was assessed as per previously reported methods [16]. Symptomatic COVID-19 was defined according to US Food and Drug Administration criteria [19]. Symptoms of suspected COVID-19 were monitored throughout the trial and collected using an electronic symptom diary for at least 10 days after symptom start date. Virological confirmation was performed using polymerase chain reaction (PCR) testing. Asymptomatic infection was defined as occurring in participants with a positive PCR test result for SARS-CoV-2 or who seroconverted after day 35 (2 weeks after second vaccination) to immunoglobulin G (IgG) against the nucleocapsid protein (N-protein) without any symptoms or with symptoms that did not meet the symptomatic end point criteria (see protocol for details; Supplementary Methods). An additional efficacy end point included the first occurrence of laboratory-confirmed (by PCR or N-protein serology test) symptomatic or asymptomatic COVID-19 with onset at least 7 days after second study vaccination in participants with negative serostatus at baseline.
Immunogenicity Assessments
Detection of SARS-CoV-2 anti–N-protein IgG (Roche Elecsys Anti-SARS-CoV-2, Indianapolis, IN) was performed at baseline, day 35, 3 months, and just before receiving a crossover dose in all participants to establish serostatus and for assessment of asymptomatic disease. An enzyme-linked immunosorbent assay (ELISA) for SARS-CoV-2 anti-S protein IgG (Novavax, Gaithersburg, MD) and a microneutralization assay (360 bioLabs, Melbourne, Australia) were performed at baseline and on day 35 in approximately 900 consecutive participants from 2 study sites (immunogenicity cohort). Induction of S-protein–specific T-cell responses by immunization was measured at baseline and on day 35 in approximately 450 consecutive participants from 2 study sites using ELISpot assays to detect T cells in peripheral blood responsive to SARS-CoV-2 S-protein peptides (Oxford Immunotech; assays are detailed in the Supplementary Methods).
Statistical Analyses
Safety Analysis
Safety events were summarized descriptively. AEs were coded by preferred term and system organ class using version 23.1 of the Medical Dictionary for Regulatory Activities and summarized by severity and relationship to study vaccine.
For participants who enrolled in the blinded crossover, safety data were censored at the time of crossover (eg, the date at which participants received their third study dose). For participants who did not enter the blinded crossover and were unblinded (but did not withdraw from the study), safety data were censored at the time of unblinding or the time of receipt of another COVID-19 vaccine (whichever date was noted first). For participants who neither entered the blinded crossover nor were censored, the safety data cutoff date was 27 July 2021 (Figure 1).
Efficacy Analysis
Efficacy analysis was conducted in the same manner as previously described [16] and is detailed in the Supplementary Methods. Participants were censored at the earliest of the date of unblinding (for any reason), date of receipt of another COVID-19 vaccine, date of entering the blinded crossover, date of early withdrawal, date of death, or the cutoff date of 27 July 2021.
Immunogenicity Analysis
For the SARS-CoV-2 anti–S-protein IgG antibody levels measured by ELISA, the geometric mean at each study visit (baseline and day 35), the geometric mean fold-rises (GMFRs) comparing day 0 (baseline) to day 35, along with 95% confidence interval (CI), were summarized. The 95% CI was calculated based on the t-distribution of the log-transformed values for geometric means or GMFRs, then backtransformed to the original scale for presentation. The seroconversion rate (SCR), proportion of participants with ≥ 4-fold rises if seronegative at baseline, and 95% CIs based on the Clopper–Pearson method are summarized by vaccine group. A similar statistical analysis was performed for the microneutralization assay.
T-cell responses to SARS-CoV-2 protein peptide pools were assessed based on counts of cells secreting interferon-gamma (IFN-γ) per 2.5 × 105 peripheral blood mononuclear cells (Oxford Immunotec, Abingdon, Oxfordshire, UK) before immunization (day 0) and at day 35. Mean spot counts (with standard deviations) were calculated by treatment group, age stratum, stimulation condition, and time point. In addition, GMFRs from baseline by treatment group and age stratum were calculated from within-participant ratios of day 35 to day 0 counts.
RESULTS
Participants
Between 28 September 2020 and 28 November 2020, 16 631 participants were screened and 15 185 participants were randomized (Figure 1). A total of 15 138 participants received at least 1 dose of NVX-CoV2373 (7569) or placebo (7569). The protocol-specified number of events for the primary event-driven analysis was reached just before the data cutoff date of 29 January 2021 and vaccine efficacy and safety have been reported [16].
The current analysis contains 158 participants who met the primary end point definition (24 in the vaccine arm and 134 in the placebo arm), occurring between 10 November 2020 and 10 May 2021. The current analysis had a maximum observation period of 7.5 months (28 September 2020 through 10 May 2021) with a median of 4.5 months. Baseline demographics for per-protocol populations are listed in Table 1. Baseline participant demographics for the immunogenicity and cell-mediated immunity cohorts are listed in Supplementary Table 3.
Demographics and Baseline Characteristics for Participants in the Per-Protocol Efficacy Analysis Set
| Parameters | NVX-CoV2373 (n = 6989) | Placebo (n = 7000) | Total (N = 13 989) |
|---|---|---|---|
| Age, y | |||
| ȃn | 6989 | 7000 | 13 989 |
| ȃMean (SD) | 53.4 (14.81) | 53.4 (14.83) | 53.4 (14.82) |
| ȃMedian | 56.0 | 56.0 | 56.0 |
| Age group, y | |||
| ȃ<65 | 5046 (72.2) | 5050 (72.1) | 10 096 (72.2) |
| ȃ≥65 | 1943 (27.8) | 1950 (27.9) | 3893 (27.8) |
| Sex | |||
| ȃMale | 3594 (51.4) | 3616 (51.7) | 7210 (51.5) |
| ȃFemale | 3395 (48.6) | 3384 (48.3) | 6779 (48.5) |
| Racea | |||
| ȃWhite | 6637 (95.0) | 6650 (95.0) | 13 287 (95.0) |
| ȃBlack or African American | 26 (0.4) | 26 (0.4) | 52 (0.4) |
| ȃAsian | 207 (3.0) | 217 (3.1) | 424 (3.0) |
| ȃMultiple | 32 (0.5) | 28 (0.4) | 60 (0.4) |
| ȃNot reported | 75 (1.1) | 69 (1.0) | 144 (1.0) |
| ȃOther | 12 (0.2) | 10 (0.1) | 22 (0.2) |
| Ethnicitya | |||
| ȃHispanic or Latino | 64 (0.9) | 51 (0.7) | 115 (0.8) |
| ȃNot Hispanic or Latino | 6268 (89.7) | 6308 (90.1) | 12 576 (89.9) |
| ȃNot reported | 537 (7.7) | 513 (7.3) | 1050 (7.5) |
| ȃUnknown | 120 (1.7) | 127 (1.8) | 247 (1.8) |
| ȃMissing | 0 | 1 | 1 |
| Baseline body mass index,b kg/m2 | |||
| ȃn | 6836 | 6847 | 13 683 |
| ȃMean (SD) | 27.52 (5.321) | 27.71 (5.637) | 27.62 (5.482) |
| ȃMedian | 26.70 | 26.80 | 26.70 |
| ȃMin, Max | 14.2, 61.2 | 10.3, 87.4 | 10.3, 87.4 |
| Baseline PCR | |||
| ȃPositive | 0 | 0 | 0 |
| ȃNegative | 6624 (94.8) | 6617 (94.5) | 13 241 (94.7) |
| ȃMissing | 365 | 383 | 748 |
| Day 21 PCRc | |||
| ȃPositive | 1 (< 0.1) | 1 (< 0.1) | 2 (< 0.1) |
| ȃNegative | 372 (5.3) | 360 (5.1) | 732 (5.2) |
| Comorbidity statusd | |||
| ȃYes | 3137 (44.9) | 3165 (45.2) | 6302 (45.0) |
| ȃNo | 3852 (55.1) | 3835 (54.8) | 7687 (55.0) |
| Parameters | NVX-CoV2373 (n = 6989) | Placebo (n = 7000) | Total (N = 13 989) |
|---|---|---|---|
| Age, y | |||
| ȃn | 6989 | 7000 | 13 989 |
| ȃMean (SD) | 53.4 (14.81) | 53.4 (14.83) | 53.4 (14.82) |
| ȃMedian | 56.0 | 56.0 | 56.0 |
| Age group, y | |||
| ȃ<65 | 5046 (72.2) | 5050 (72.1) | 10 096 (72.2) |
| ȃ≥65 | 1943 (27.8) | 1950 (27.9) | 3893 (27.8) |
| Sex | |||
| ȃMale | 3594 (51.4) | 3616 (51.7) | 7210 (51.5) |
| ȃFemale | 3395 (48.6) | 3384 (48.3) | 6779 (48.5) |
| Racea | |||
| ȃWhite | 6637 (95.0) | 6650 (95.0) | 13 287 (95.0) |
| ȃBlack or African American | 26 (0.4) | 26 (0.4) | 52 (0.4) |
| ȃAsian | 207 (3.0) | 217 (3.1) | 424 (3.0) |
| ȃMultiple | 32 (0.5) | 28 (0.4) | 60 (0.4) |
| ȃNot reported | 75 (1.1) | 69 (1.0) | 144 (1.0) |
| ȃOther | 12 (0.2) | 10 (0.1) | 22 (0.2) |
| Ethnicitya | |||
| ȃHispanic or Latino | 64 (0.9) | 51 (0.7) | 115 (0.8) |
| ȃNot Hispanic or Latino | 6268 (89.7) | 6308 (90.1) | 12 576 (89.9) |
| ȃNot reported | 537 (7.7) | 513 (7.3) | 1050 (7.5) |
| ȃUnknown | 120 (1.7) | 127 (1.8) | 247 (1.8) |
| ȃMissing | 0 | 1 | 1 |
| Baseline body mass index,b kg/m2 | |||
| ȃn | 6836 | 6847 | 13 683 |
| ȃMean (SD) | 27.52 (5.321) | 27.71 (5.637) | 27.62 (5.482) |
| ȃMedian | 26.70 | 26.80 | 26.70 |
| ȃMin, Max | 14.2, 61.2 | 10.3, 87.4 | 10.3, 87.4 |
| Baseline PCR | |||
| ȃPositive | 0 | 0 | 0 |
| ȃNegative | 6624 (94.8) | 6617 (94.5) | 13 241 (94.7) |
| ȃMissing | 365 | 383 | 748 |
| Day 21 PCRc | |||
| ȃPositive | 1 (< 0.1) | 1 (< 0.1) | 2 (< 0.1) |
| ȃNegative | 372 (5.3) | 360 (5.1) | 732 (5.2) |
| Comorbidity statusd | |||
| ȃYes | 3137 (44.9) | 3165 (45.2) | 6302 (45.0) |
| ȃNo | 3852 (55.1) | 3835 (54.8) | 7687 (55.0) |
Data are n (%) unless otherwise indicated. Percentages are based on per-protocol efficacy analysis set within each treatment and overall.
Abbreviations: PCR, polymerase chain reaction; SD, standard deviation.
Race or ethnic group was reported by the participants who could have listed more than 1 category.
Body mass index is calculated as weight (kg) divided by squared height (m). A value >30 kg/m2 is considered to indicate obesity.
Test performed only if the participant has any coronavirus disease 2019 (COVID-19) symptoms or significant exposure history between days 0 and 21.
Comorbid participants are those identified who have at least 1 of the comorbid conditions reported as a medical history or have a screening body mass index value >30 kg/m2. Coexisting conditions were recognized risk factors for severe COVID-19. These included chronic respiratory, cardiac, renal, neurologic, hepatic, and certain immunocompromising conditions, as well as obesity.
Demographics and Baseline Characteristics for Participants in the Per-Protocol Efficacy Analysis Set
| Parameters | NVX-CoV2373 (n = 6989) | Placebo (n = 7000) | Total (N = 13 989) |
|---|---|---|---|
| Age, y | |||
| ȃn | 6989 | 7000 | 13 989 |
| ȃMean (SD) | 53.4 (14.81) | 53.4 (14.83) | 53.4 (14.82) |
| ȃMedian | 56.0 | 56.0 | 56.0 |
| Age group, y | |||
| ȃ<65 | 5046 (72.2) | 5050 (72.1) | 10 096 (72.2) |
| ȃ≥65 | 1943 (27.8) | 1950 (27.9) | 3893 (27.8) |
| Sex | |||
| ȃMale | 3594 (51.4) | 3616 (51.7) | 7210 (51.5) |
| ȃFemale | 3395 (48.6) | 3384 (48.3) | 6779 (48.5) |
| Racea | |||
| ȃWhite | 6637 (95.0) | 6650 (95.0) | 13 287 (95.0) |
| ȃBlack or African American | 26 (0.4) | 26 (0.4) | 52 (0.4) |
| ȃAsian | 207 (3.0) | 217 (3.1) | 424 (3.0) |
| ȃMultiple | 32 (0.5) | 28 (0.4) | 60 (0.4) |
| ȃNot reported | 75 (1.1) | 69 (1.0) | 144 (1.0) |
| ȃOther | 12 (0.2) | 10 (0.1) | 22 (0.2) |
| Ethnicitya | |||
| ȃHispanic or Latino | 64 (0.9) | 51 (0.7) | 115 (0.8) |
| ȃNot Hispanic or Latino | 6268 (89.7) | 6308 (90.1) | 12 576 (89.9) |
| ȃNot reported | 537 (7.7) | 513 (7.3) | 1050 (7.5) |
| ȃUnknown | 120 (1.7) | 127 (1.8) | 247 (1.8) |
| ȃMissing | 0 | 1 | 1 |
| Baseline body mass index,b kg/m2 | |||
| ȃn | 6836 | 6847 | 13 683 |
| ȃMean (SD) | 27.52 (5.321) | 27.71 (5.637) | 27.62 (5.482) |
| ȃMedian | 26.70 | 26.80 | 26.70 |
| ȃMin, Max | 14.2, 61.2 | 10.3, 87.4 | 10.3, 87.4 |
| Baseline PCR | |||
| ȃPositive | 0 | 0 | 0 |
| ȃNegative | 6624 (94.8) | 6617 (94.5) | 13 241 (94.7) |
| ȃMissing | 365 | 383 | 748 |
| Day 21 PCRc | |||
| ȃPositive | 1 (< 0.1) | 1 (< 0.1) | 2 (< 0.1) |
| ȃNegative | 372 (5.3) | 360 (5.1) | 732 (5.2) |
| Comorbidity statusd | |||
| ȃYes | 3137 (44.9) | 3165 (45.2) | 6302 (45.0) |
| ȃNo | 3852 (55.1) | 3835 (54.8) | 7687 (55.0) |
| Parameters | NVX-CoV2373 (n = 6989) | Placebo (n = 7000) | Total (N = 13 989) |
|---|---|---|---|
| Age, y | |||
| ȃn | 6989 | 7000 | 13 989 |
| ȃMean (SD) | 53.4 (14.81) | 53.4 (14.83) | 53.4 (14.82) |
| ȃMedian | 56.0 | 56.0 | 56.0 |
| Age group, y | |||
| ȃ<65 | 5046 (72.2) | 5050 (72.1) | 10 096 (72.2) |
| ȃ≥65 | 1943 (27.8) | 1950 (27.9) | 3893 (27.8) |
| Sex | |||
| ȃMale | 3594 (51.4) | 3616 (51.7) | 7210 (51.5) |
| ȃFemale | 3395 (48.6) | 3384 (48.3) | 6779 (48.5) |
| Racea | |||
| ȃWhite | 6637 (95.0) | 6650 (95.0) | 13 287 (95.0) |
| ȃBlack or African American | 26 (0.4) | 26 (0.4) | 52 (0.4) |
| ȃAsian | 207 (3.0) | 217 (3.1) | 424 (3.0) |
| ȃMultiple | 32 (0.5) | 28 (0.4) | 60 (0.4) |
| ȃNot reported | 75 (1.1) | 69 (1.0) | 144 (1.0) |
| ȃOther | 12 (0.2) | 10 (0.1) | 22 (0.2) |
| Ethnicitya | |||
| ȃHispanic or Latino | 64 (0.9) | 51 (0.7) | 115 (0.8) |
| ȃNot Hispanic or Latino | 6268 (89.7) | 6308 (90.1) | 12 576 (89.9) |
| ȃNot reported | 537 (7.7) | 513 (7.3) | 1050 (7.5) |
| ȃUnknown | 120 (1.7) | 127 (1.8) | 247 (1.8) |
| ȃMissing | 0 | 1 | 1 |
| Baseline body mass index,b kg/m2 | |||
| ȃn | 6836 | 6847 | 13 683 |
| ȃMean (SD) | 27.52 (5.321) | 27.71 (5.637) | 27.62 (5.482) |
| ȃMedian | 26.70 | 26.80 | 26.70 |
| ȃMin, Max | 14.2, 61.2 | 10.3, 87.4 | 10.3, 87.4 |
| Baseline PCR | |||
| ȃPositive | 0 | 0 | 0 |
| ȃNegative | 6624 (94.8) | 6617 (94.5) | 13 241 (94.7) |
| ȃMissing | 365 | 383 | 748 |
| Day 21 PCRc | |||
| ȃPositive | 1 (< 0.1) | 1 (< 0.1) | 2 (< 0.1) |
| ȃNegative | 372 (5.3) | 360 (5.1) | 732 (5.2) |
| Comorbidity statusd | |||
| ȃYes | 3137 (44.9) | 3165 (45.2) | 6302 (45.0) |
| ȃNo | 3852 (55.1) | 3835 (54.8) | 7687 (55.0) |
Data are n (%) unless otherwise indicated. Percentages are based on per-protocol efficacy analysis set within each treatment and overall.
Abbreviations: PCR, polymerase chain reaction; SD, standard deviation.
Race or ethnic group was reported by the participants who could have listed more than 1 category.
Body mass index is calculated as weight (kg) divided by squared height (m). A value >30 kg/m2 is considered to indicate obesity.
Test performed only if the participant has any coronavirus disease 2019 (COVID-19) symptoms or significant exposure history between days 0 and 21.
Comorbid participants are those identified who have at least 1 of the comorbid conditions reported as a medical history or have a screening body mass index value >30 kg/m2. Coexisting conditions were recognized risk factors for severe COVID-19. These included chronic respiratory, cardiac, renal, neurologic, hepatic, and certain immunocompromising conditions, as well as obesity.
Safety
All 15 138 participants who received at least 1 dose of vaccine or placebo were assessed for safety events. NVX-CoV2373 recipients reported higher frequencies of solicited local and systemic AEs than placebo recipients after both the first dose and the second dose, with most events being mild to moderate in severity and of short mean duration [16]. The frequency of unsolicited AEs was higher among NVX-CoV2373 recipients than among placebo recipients (27.4% vs 21.8%), with similar frequencies of severe AEs, SAEs, MAAEs, AEs leading to dose or study discontinuation, potential immune-mediated medical conditions, and AESIs relevant to COVID-19 (Table 2, Supplementary Table 4).
Overall Summary of Unsolicited Adverse Events for the Safety Population
| Parameters | NVX-CoV2373 (n = 7569) | Placebo (n = 7569) | ||
|---|---|---|---|---|
| n (%) | N | n (%) | N | |
| Any AEs | 2075 (27.4) | 3134 | 1649 (21.8) | 2577 |
| Any severe AEs | 88 (1.2) | 114 | 87 (1.1) | 113 |
| Any treatment-related AEs | 880 (11.6) | 1145 | 369 (4.9) | 489 |
| Any severe, treatment-related AEs | 15 (0.2) | 17 | 5 (<0.1) | 5 |
| Any MAAEs | 355 (4.7) | 419 | 336 (4.4) | 402 |
| Any treatment-related MAAEs | 36 (0.5) | 46 | 17 (0.2) | 19 |
| Any serious AEs | 59 (0.8) | 75 | 61 (0.8) | 72 |
| Any AEs leading to vaccination discontinuation | 23 (0.3) | 29 | 28 (0.4) | 50 |
| Any treatment-related AEs leading to vaccination discontinuation | 7 (<0.1) | 11 | 8 (0.1) | 9 |
| Any AEs leading to study discontinuation | 18 (0.2) | 18 | 13 (0.2) | 13 |
| Any treatment-related AEs leading to study discontinuation | 3 (<0.1) | 3 | 1 (<0.1) | 1 |
| Any potential immune-mediated medical conditions | 6 (<0.1) | 6 | 9 (0.1) | 9 |
| Any adverse event of special interest relevant to coronavirus disease 2019 | 12 (0.2) | 18 | 35 (0.5) | 51 |
| Parameters | NVX-CoV2373 (n = 7569) | Placebo (n = 7569) | ||
|---|---|---|---|---|
| n (%) | N | n (%) | N | |
| Any AEs | 2075 (27.4) | 3134 | 1649 (21.8) | 2577 |
| Any severe AEs | 88 (1.2) | 114 | 87 (1.1) | 113 |
| Any treatment-related AEs | 880 (11.6) | 1145 | 369 (4.9) | 489 |
| Any severe, treatment-related AEs | 15 (0.2) | 17 | 5 (<0.1) | 5 |
| Any MAAEs | 355 (4.7) | 419 | 336 (4.4) | 402 |
| Any treatment-related MAAEs | 36 (0.5) | 46 | 17 (0.2) | 19 |
| Any serious AEs | 59 (0.8) | 75 | 61 (0.8) | 72 |
| Any AEs leading to vaccination discontinuation | 23 (0.3) | 29 | 28 (0.4) | 50 |
| Any treatment-related AEs leading to vaccination discontinuation | 7 (<0.1) | 11 | 8 (0.1) | 9 |
| Any AEs leading to study discontinuation | 18 (0.2) | 18 | 13 (0.2) | 13 |
| Any treatment-related AEs leading to study discontinuation | 3 (<0.1) | 3 | 1 (<0.1) | 1 |
| Any potential immune-mediated medical conditions | 6 (<0.1) | 6 | 9 (0.1) | 9 |
| Any adverse event of special interest relevant to coronavirus disease 2019 | 12 (0.2) | 18 | 35 (0.5) | 51 |
All counts exclude reactogenicity AEs (selected preferred terms). Unsolicited AEs were classified as severe, medically attended, serious, leading to vaccination or study discontinuation, potential immune-mediated medical conditions, or adverse event of special interest.
Abbreviations: AE, adverse event; MAAE, medically attended adverse event.
Overall Summary of Unsolicited Adverse Events for the Safety Population
| Parameters | NVX-CoV2373 (n = 7569) | Placebo (n = 7569) | ||
|---|---|---|---|---|
| n (%) | N | n (%) | N | |
| Any AEs | 2075 (27.4) | 3134 | 1649 (21.8) | 2577 |
| Any severe AEs | 88 (1.2) | 114 | 87 (1.1) | 113 |
| Any treatment-related AEs | 880 (11.6) | 1145 | 369 (4.9) | 489 |
| Any severe, treatment-related AEs | 15 (0.2) | 17 | 5 (<0.1) | 5 |
| Any MAAEs | 355 (4.7) | 419 | 336 (4.4) | 402 |
| Any treatment-related MAAEs | 36 (0.5) | 46 | 17 (0.2) | 19 |
| Any serious AEs | 59 (0.8) | 75 | 61 (0.8) | 72 |
| Any AEs leading to vaccination discontinuation | 23 (0.3) | 29 | 28 (0.4) | 50 |
| Any treatment-related AEs leading to vaccination discontinuation | 7 (<0.1) | 11 | 8 (0.1) | 9 |
| Any AEs leading to study discontinuation | 18 (0.2) | 18 | 13 (0.2) | 13 |
| Any treatment-related AEs leading to study discontinuation | 3 (<0.1) | 3 | 1 (<0.1) | 1 |
| Any potential immune-mediated medical conditions | 6 (<0.1) | 6 | 9 (0.1) | 9 |
| Any adverse event of special interest relevant to coronavirus disease 2019 | 12 (0.2) | 18 | 35 (0.5) | 51 |
| Parameters | NVX-CoV2373 (n = 7569) | Placebo (n = 7569) | ||
|---|---|---|---|---|
| n (%) | N | n (%) | N | |
| Any AEs | 2075 (27.4) | 3134 | 1649 (21.8) | 2577 |
| Any severe AEs | 88 (1.2) | 114 | 87 (1.1) | 113 |
| Any treatment-related AEs | 880 (11.6) | 1145 | 369 (4.9) | 489 |
| Any severe, treatment-related AEs | 15 (0.2) | 17 | 5 (<0.1) | 5 |
| Any MAAEs | 355 (4.7) | 419 | 336 (4.4) | 402 |
| Any treatment-related MAAEs | 36 (0.5) | 46 | 17 (0.2) | 19 |
| Any serious AEs | 59 (0.8) | 75 | 61 (0.8) | 72 |
| Any AEs leading to vaccination discontinuation | 23 (0.3) | 29 | 28 (0.4) | 50 |
| Any treatment-related AEs leading to vaccination discontinuation | 7 (<0.1) | 11 | 8 (0.1) | 9 |
| Any AEs leading to study discontinuation | 18 (0.2) | 18 | 13 (0.2) | 13 |
| Any treatment-related AEs leading to study discontinuation | 3 (<0.1) | 3 | 1 (<0.1) | 1 |
| Any potential immune-mediated medical conditions | 6 (<0.1) | 6 | 9 (0.1) | 9 |
| Any adverse event of special interest relevant to coronavirus disease 2019 | 12 (0.2) | 18 | 35 (0.5) | 51 |
All counts exclude reactogenicity AEs (selected preferred terms). Unsolicited AEs were classified as severe, medically attended, serious, leading to vaccination or study discontinuation, potential immune-mediated medical conditions, or adverse event of special interest.
Abbreviations: AE, adverse event; MAAE, medically attended adverse event.
There were no episodes of anaphylaxis, thrombosis with thrombocytopenia syndrome, or evidence of vaccine-associated enhanced COVID-19. There was 1 episode of myocarditis previously reported [16] and no cases of pericarditis. By the end of the placebo-controlled phase, there were 7 deaths (NVX-Co2373, n = 4; placebo, n = 3), none of which were considered related to study vaccine.
Efficacy
Among 13 831 participants in the per-protocol efficacy population before the blinded crossover, there were 24 cases of virologically (PCR) confirmed, symptomatic mild, moderate, or severe COVID-19 with onset at least 7 days after the second dose among vaccine recipients (9.48 per 1000 person-years; 95% CI, 5.82–15.42) and 134 cases among placebo recipients (54.85 per 1000 person-years; 95% CI, 41.03–73.32) for a vaccine efficacy of 82.7% (95% CI, 73.3%–88.8%). Vaccine efficacy against moderate or severe disease was 79.2% (95% CI, 66.7%–87.0%), and efficacy against severe COVID-19 was 100% (95% CI, 17.9%–100.0%); all 6 participants with severe COVID-19 had received placebo (Figure 2).
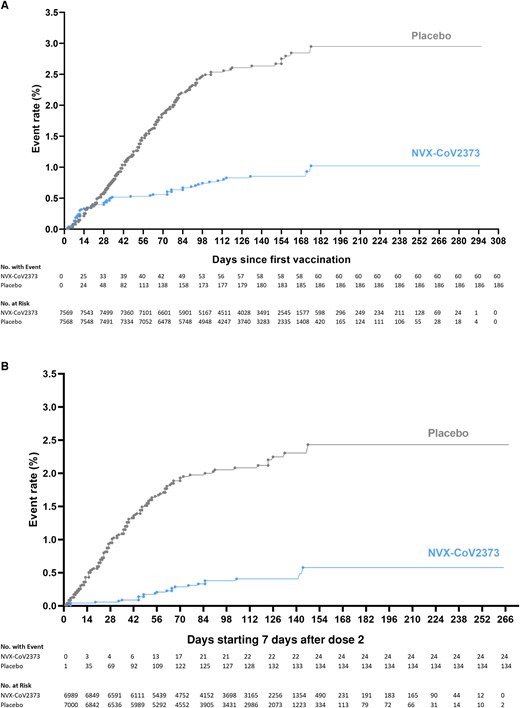
Kaplan–Meier plots of efficacy of NVX-CoV2373 against symptomatic coronavirus disease 2019 (COVID-19) in the per-protocol and intention-to-treat analysis sets. Cumulative incidence of symptomatic COVID-19 in the intent-to-treat population (A) and the per-protocol population (B). The timing of surveillance for symptomatic COVID-19 began after the first dose (intention-to-treat population) and at least 7 days after the administration of the second dose (per-protocol population) of vaccine or placebo (ie, on day 28) through a median of approximately 4.5 months of follow-up.
Additional efficacy analyses (among subgroups defined by age, race, sex, presence of comorbid conditions, influenza vaccine coadministration, and disease severity) are detailed in Figure 3.
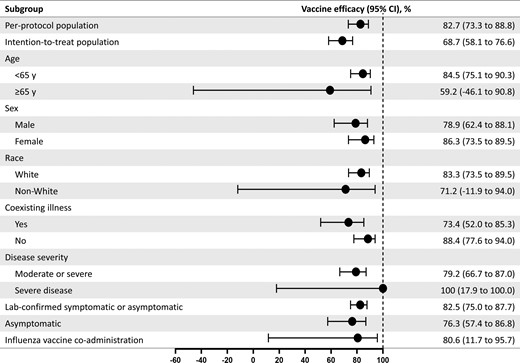
Vaccine efficacy of NVX-CoV2373 in specific subgroups. The efficacy of NVX-CoV2373 in preventing coronavirus disease 2019 (COVID-19) in various subgroups within the per-protocol population. Vaccine efficacy was defined as 1 minus the relative risk (NVX-CoV2373 vs placebo), and 95% confidence intervals were derived using Poisson regression with robust error variance (except where noted when the Clopper–Pearson exact binomial method was used). Vaccine efficacy for the intention-to-treat population was assessed after dose 1. Data in non-White populations consisted of minority and multiple races, which were pooled to ensure that the subpopulations would be large enough for meaningful analyses. Comorbidity assessment is based on the Centers for Disease Control and Prevention definition of those at increased risk for COVID-19. The laboratory-confirmed symptomatic or asymptomatic and asymptomatic end points are defined in the text. Influenza vaccine coadministration was assessed as part of a predefined influenza vaccine coadministration substudy. Abbreviation: CI, confidence interval.
A total of 70 participants (NVX-CoV2373, n = 14; placebo, n = 56) were found to have asymptomatic infection, a vaccine efficacy of 76.3% (95% CI, 57.4%–86.8%). Finally, 231 participants (NVX-CoV2373, n = 36; placebo, n = 195) had either symptomatic or asymptomatic infection, a vaccine efficacy of 82.5% (95% CI, 75.0%–87.7%).
Immunogenicity
The per-protocol anti–S-protein serology subset was composed of 831 participants (NVX-CoV2373, n = 414; placebo, n = 417), and the per-protocol neutralization assay subset included 761 participants (NVX-CoV2373, n = 381; placebo, n = 380). The immunology sets were well balanced between the 2 vaccine study groups (Supplementary Tables 5–11).
Serum anti–S-protein IgG levels in participants at day 35 were increased relative to placebo across all age groups (Supplementary Table 5, Supplementary Figures 1–4). Serum anti–S-protein IgG geometric mean ELISA units (EU)/mL (GMCs) in the NVX-CoV2373 group were highest in the younger vs older age cohort (18–64 years, 47 564.3 EU/mL; 65–84 years, 37 892.8 EU/mL). Similar differences in responses were observed regardless of baseline serostatus (Supplementary Tables 5–7). Serum anti–S-protein IgG GMCs in the NVX-CoV2373 group were highest in the seropositive cohort (125 489.8 EU/mL) vs the seronegative cohort (44 229.9 EU/mL; Supplementary Table 7). These responses equated to serum anti–S-protein IgG GMFRs relative to baseline of 73.9 and 394.3, respectively. SCRs were markedly increased relative to placebo across all baseline serostatus groups (98.9% for all participants; 95.7% seropositive and 99.1% seronegative) and regardless of age group (18–64 years, 99.0%; 65–84 years, 99.1%).
A similar pattern was observed with neutralizing antibody responses, with the highest responses seen in the younger age cohort (Supplementary Tables 8–10, Supplementary Figures 5–11) and in those who were seropositive at baseline (Supplementary Table 10, Supplementary Figure 11).
T-Cell Responses
ELISpot assays were performed on peripheral blood from 407 per-protocol participants (Supplementary Table 11, Supplementary Figure 12). In the NVX-CoV2373 group, strong induction of T cells secreting IFN-γ occurred in response to peptide pools reflecting the full length of the SARS-CoV-2 S-protein, and to its N-terminal and C-terminal portions, with GMFRs of 16.5-, 14.2-, and 8.4-fold, respectively. C-terminal sequences elicited somewhat lesser responses than the full-length or N-terminal peptide pools but with a similar pattern. The amplitude of T-cell responses was generally lower in vaccinated participants aged ≥ 65 years.
DISCUSSION
The data through to the completion of the placebo-controlled stage of this phase 3 trial provide further evidence of the safety and efficacy of NVX-CoV2373 in preventing symptomatic COVID-19. These findings are based on >6 months (median, 4.5) of follow-up and are similar to those observed previously at a median follow-up of 3 months [16], indicating only a small reduction in NVX-CoV2373 vaccine efficacy. Of note, NVX-CoV2373 provided substantial protection from laboratory-confirmed asymptomatic infection (76.3%; 95% CI, 57.4%–86.8%), higher than that reported from randomized controlled trials of other COVID-19 vaccines [11–13] and provided a combined vaccine efficacy of 82.5% (95% CI, 75.0%–87.7%) against laboratory-confirmed symptomatic or asymptomatic infection. Prevention of both symptomatic and asymptomatic infection is of paramount importance for interrupting viral transmission.
A 2-dose regimen of NVX-CoV2373, administered 21 days apart, markedly increased anti–S-protein IgG and neutralizing antibody levels, regardless of baseline serostatus, with higher levels in the younger vs older adult cohort and in those who were seropositive at baseline. Similar differences in responses related to age and prior exposure have been described for other COVID-19 vaccines and are consistent with immunosenescence and priming, respectively [20, 21]. There was a high correlation between “binding” antibodies (anti–S-protein IgG) and neutralizing antibodies, indicating that the anti–S-protein immunity was predominantly functional. Additionally, evaluation of T-cell responses demonstrates induction of a T-cell population that secretes IFN-γ in response to epitopes within the SARS-CoV-2 S-protein.
The small reduction in vaccine efficacy over time may relate to a decrease in neutralizing antibody titers, as reported for this [22] and other COVID-19 vaccines [23, 24]. To maintain high levels of efficacy, especially for those populations with the greatest susceptibility to severe disease, a booster dose may be warranted. Booster vaccines are most likely to be of greatest importance for those aged ≥65 years who exhibited lower titers and a more significant decrease in vaccine efficacy than those aged <65 years. In another study, a single dose of NVX-CoV2373 administered at 6 months after primary vaccination with 2 doses of NVX-CoV2373 resulted in a marked increase in titers against the prototype strain and all variants evaluated, notably greater than those titers associated with the high levels of efficacy in phase 3 studies of this vaccine [22]. A separate study found significantly improved neutralization of SARS-CoV-2 variants Omicron BA.1 (35-fold increase; P < .001) and BA.4/BA.5 (12-fold increase; P < .001) after a third (booster) dose of NVX-CoV2373 [25]. A study with NVX-CoV2373 administered after a primary series of either the ChAdOx1nCoV-19 or BNT162b2 vaccines showed boosted antibody and neutralizing responses with low levels of reactogenicity [26]. Additional homologous and heterologous studies of NVX-CoV2373 as a booster dose are underway in different populations [6, 17, 22, 26, 27].
The favorable safety profile observed earlier in this study was found to be consistent through to the end of the placebo-controlled period. The incidence of SAEs was similar in the vaccine and placebo groups, and no deaths were attributable to receipt of the vaccine. To date, the findings of this and other studies assessing the safety of NVX-CoV2373 [6, 16, 17] have shown no evidence of increased risk for myocarditis/pericarditis or thrombocytopenic–thrombotic events.
This trial has several limitations. The overall duration of the placebo-controlled period was short but necessary to offer study participants the option of receiving the active vaccine. Sequencing data on study isolates were not available, although contemporary data from national surveillance shows the dominance of the Alpha variant during the study period together with the emergence of the Delta variant (Figure 4) [28]. As both variants have shown lower vaccine efficacy point estimates with NVX-CoV2373 than against the earlier Wuhan strain [16, 17, 29], this shifting variant landscape, along with the additional time since vaccination, may have contributed to a decrease in vaccine efficacy.
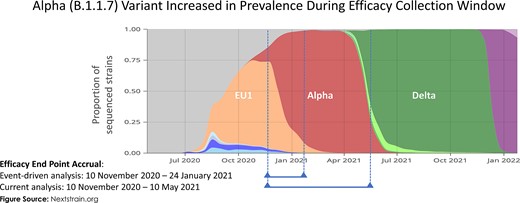
Emergence of the Alpha and Delta variants during the study assessment periods in the United Kingdom. Surveillance data are presented on the severe acute respiratory syndrome coronavirus 2 strains that were present during the initial event-driven analysis and the current analysis periods. The Alpha variant emerged during the event-driven analysis and was present in more than half of the end points assessed at that time. In the subsequent time until the end of the placebo-controlled period, the Alpha variant became the dominant variant and the United Kingdom saw the beginning of the emergence of the Delta variant.
The results of this trial provide further evidence that symptomatic, asymptomatic, and severe SARS-CoV-2 infections can be prevented by a protein-based, adjuvanted vaccine within a maximal period of 7.5 months. NVX-CoV2373 has received conditional authorization in many locations, and this will allow for the accumulation of real-world evidence to further assess vaccine effectiveness, safety, and duration of protection.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Author contributions. P. T. H. is the chief investigator. P. T. H., S. T., G. G., I. C., and A. R. contributed to the protocol and design of the study. S. T. and P. T. H. contributed to the design and execution of the study; the analysis and interpretation of the data; and the writing, reviewing, and editing of text and figures. E. P. G., D. N. B., M. B., D. B., F. B., D. R. C., R. C., C. A. C., J. G., A. L. G., A. He., A. Hi., S. I., C. J., P. A. K., C. K., J. M. B., C. M., A. M. M., F. M., P. M., I. Mu., H. N., O. O., J. P., C. H. P., A. S. F. R., D. S., R. P. S., R. S., R. L. S., P. A. S., E. C. T., J. T., and M. E. V. are study site principal investigators and contributed to the study data collection. I. C. and A. R. conducted the statistical analysis. All authors reviewed, commented on, and approved the manuscript before submission for publication.
Acknowledgments. The authors thank all study participants for their commitment to this study. They also acknowledge the investigators and their study teams for their hard work and dedication. In addition, they thank the National Institute for Health Research, representatives from the Department of Health and Social Care laboratories and National Health Service (NHS) Digital, and the members of the UK Vaccine Task Force. Editorial support was provided by Kelly Cameron of Ashfield MedComms, an Inizio company.
Disclaimer. The sponsor had primary responsibility for study design, study vaccines, protocol development, study monitoring, data management, and statistical analyses.
Financial support. This work was funded by Novavax.
Data sharing. The trial protocol and statistical analysis plan have been made available as part of the peer-review process and will be made available upon publishing of the article by request to the corresponding author. Additional information is available at clinicaltrialsregister.eu (EudraCT, 2020-004123-16).
References
Author notes
Potential conflicts of interest. K. A., I. C., L. F., G. G., I. Mc., E. J. R., A. R., K. S., and S. T. are employees of Novavax Inc and receive a salary for their work. K. S. reports stock received as part of employment compensation from Novavax. A. R. reports stock or stock options from Novavax. E. J. R. and I. Mc. report Novavax stock. K. A. reports vested and unvested Novavax stock/restricted stock units. I. C. reports Novavax stock and stock options and salary and bonus as an employee of Novavax. L. F. reports consulting fees as a prior full-time employee, now contractor to, Novavax reimbursed hourly for work performed on this study and in analyses and drafting this report, and shares and stock options from Novavax. G. G. reports stock-related compensations from Novavax. S. T. reports royalties or licenses, salary and stock, payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events, and a leadership or fiduciary role in other board, society, committee, or advocacy group, paid or unpaid, as an employee of Novavax. Guy's and St Thomas' National Health Service (NHS) Foundation Trust (with which A. L. G. is affiliated) received funding from Novavax for this trial. Oxford University has entered into a partnership with AstraZeneca for further development of ChAdOx1 nCoV-19. A. L. G. is named as an inventor on a patent covering use of a particular promoter construct that is often used in vectored vaccines and is incorporated in the ChAdOx1 nCoV-19 vaccine, and may benefit from royalty income paid to the University of Oxford from sales of this vaccine by AstraZeneca and its sublicensees under the university's revenue sharing policy. M. B. reports a research grant to institution from Novavax related to this manuscript, advisory/speaker fees or grants to the institution from GSK, ViiV, Gilead, Janssen, Moderna, Pfizer, Valneva, MSD, Roche, Cipla, and Mylan and support for attending the online World AIDS conference from ViiV. D. R. C. reports a research grant to institution from Gilead Sciences, unpaid participation as an Independent Data Monitoring Board (IDMB) member for the FLARE Trial, and unpaid role as a British HIV Association trustee member. J. G. reports a research contract with institution from Novavax. C. A. C. reports a research grant to institution from Novavax related to this manuscript, and a research grant to institution from Moderna. P. T. H. reports a research grant to institution from Novavax related to this manuscript, research grants to institution from Pfizer, AstraZeneca, Moderna, Valneva, and Janssen, and payment to institution for educational events from Novavax. P. A. K. reports a research grant to institution from Novavax related to this manuscript, grants or contracts unrelated to this work from Vifor, Astellas, Evotec, Pharmacosmos, and Unicyte; consulting fees from AstraZeneca, Vifor, Unicyte, and UCB; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Vifor, AstraZeneca, Pfizer, Pharmacosmos, Napp, and Bayer; and support for attending meetings and/or travel from Pharmacosmos and Vifor. J. P. reports a research grant to institution from Novavax related to this manuscript, and being co-applicant for a Haywood Foundation grant and for a National Institute for Health and Care Research (NIHR)/Clinical Research Network (CRN) COVID Innovation and Insight grant. P. A. S. reports a research grant to institution from Novavax related to this manuscript, payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Bayer and AstraZeneca; support for attending meetings and/or travel from Bayer; and participation on a DSMB or advisory board for Bayer. E. C. T. reports a research grant to institution from Novavax related to this manuscript, research grants to institution from Valneva, COV-BOOST, Medical Research Council (MRC), Wellcome, and Public Health Scotland; payment to author for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Wellcome Connecting Science—Sanger Institute; support for attending meetings and/or travel, paid to author, from Wellcome Connecting Science—Sanger Institute, University of Oxford, University of Cambridge, and University of Manchester; and unpaid leadership or fiduciary roles with the Scottish Committee for Pandemic Preparedness, UK Health Security Agency (HSA) technical groups, and the Scottish Genomics Oversight Group. J. T. reports Quin Technologies consulting fees; participation as chair of the Data Monitoring and Ethics Committee (DMEC) for the NIFTY Trial, an NIHR-funded trial; and a role as clinical advisor to Parathyroid UK, a patient support charity. S. I., C. J., H. N., D. B., A. Hi., R. S., I. M., A. M., R. C., D. N. B., D. S., E. P. G., J. B., R. L. S., A. He., C. M., C. P., and F. B. report a research grant to institution from Novavax related to this manuscript. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}