






Abstract
In the ION-4 trial, hepatitis C virus relapse was rare, occurring only in African American patients, 80% receiving efavirenz for human immunodeficiency virus infection. We observed no indication that CYP2B6 polymorphisms associated with increased plasma efavirenz exposure explained the relapses.
New hepatitis C virus (HCV) direct-acting antiviral treatments, including the fixed-dose combination of ledipasvir/sofosbuvir (LDV/SOF), are safe and highly effective, including in populations with poorer responses to older treatments, such as individuals coinfected with HCV and human immunodeficiency virus (HIV) [1]. However, concurrent treatment for HCV and HIV-1 increases the possibility of drug-drug interactions, because one drug may induce multiple metabolizing enzymes [2, 3].
In the phase 3 ION-4 trial, 335 HIV/HCV-coinfected patients received concomitant antiretroviral (ARV) therapy and LDV/SOF for 12 weeks; 10 patients experience a relapse of HCV infection after treatment. Two factors implicated a possible genetic mechanism in these relapse events: (1) relapse occurred only in individuals of African American ancestry and (2) most patients with relapse (8 of 10) were receiving an efavirenz (EFV)–based ARV regimen. EFV is primarily metabolized by the enzymes CYP2B6 and, to a lesser extent, CYP3A4/5 [4]. Two CYP2B6 variants more common among African Americans, rs3745274 (516G → T) and rs28399499 (983T → C), are known to reduce EFV metabolism, leading to up to 5-fold increased plasma EFV concentrations [5, 6].
This seemed particularly intriguing, because phase 1 data indicated an approximately 30% reduction in plasma LDV exposure in the presence of EFV [7]. Thus, although the mechanism of LDV metabolism is unknown, we hypothesized that increased blood levels of EFV in individuals carrying the slow-metabolizing CYP2B6 allele(s) might result in rapid degradation of LDV via the induction of metabolizing enzymes, accounting for the observed treatment relapse.
Given the potential genetic component related to HCV infection relapse in the HIV/HCV-coinfected ION-4 cohort, we conducted a candidate gene investigation to determine the association of the 2 known CYP2B6 slow-metabolizer single- nucleotide polymorphisms (SNPs) with HCV relapse. We further conducted a genome-wide association study (GWAS) investigating other common genetic variants potentially related to HCV relapse in this cohort. We herein report the results of our investigation of genetic variation related to HCV relapse after LDV/SOF therapy in HIV/HCV-coinfected individuals receiving ARV therapy.
MATERIALS AND METHODS
Patient Population
ION-4 was a multicenter, open-label, phase 3 clinical trial of LDV/SOF for 12 weeks in HIV/HCV-coinfected subjects. Of the 335 patients in ION-4 who consented to genetic testing, 278 had available DNA for this study. The ION-4 cohort has been described elsewhere [1]. Briefly, patients were ≥18 years of age, coinfected with HIV and HCV, and receiving a suppressive ARV regimen (emtricitabine and tenofovir disoproxil fumarate plus EFV, raltegravir, or rilpivirine). Patients were predominantly male (82%) and infected with HCV genotype 1a (75%). Thirty-four percent were African American, and 48% were receiving an EFV-containing ARV regimen. Robust EFV and LDV pharmacokinetic data were not available. Patients who consented to genetic testing (n = 335) were genotyped for IL28B (rs12979860). Other causes of treatment failure were explored in ION-4, but those findings did not explain the observed relapses investigated in the current study [1].
Genotyping
Of the 278 patient samples, 275 were available for GWAS and 267 for TaqMan genotyping, including samples from 7 of the 10 patients with treatment relapse, 6 of whom were receiving EFV, as well as 1 treatment nonresponder not receiving EFV. TaqMan genotyping was performed on the CYP2B6 SNPs rs3745274 and rs28399499 using the Applied Biosystems TaqMan SNP Genotyping Assay on an Applied Biosystems 7900HT fast real-time polymerase chain reaction system. The rs28399499 SNP was manually called owing to its low minor allele frequency (MAF), because no homozygous variant individuals were observed.
After manual calling, the frequencies for rs28399499 in the genotyped cohort (MAF, 5.81% in African Americans and 0.41% in whites) were similar to expected frequencies for the dominant populations in publicly available databases [8]. For rs3745274, the MAF was 43.53% (population MAF, 37%) for African Americans and 22.76% (population MAF, 24%) for whites [8]. Of the 267 individuals genotyped with the TaqMan system, 166 were white, 86 African American, and 15 categorized as other or unknown. Genotypes were successfully called in 265 individuals for rs3745274 and 267 individuals for rs28399499, including all 7 available patients with treatment relapse (6 receiving EFV).
GWAS genotyping was performed on Illumina HumanCore chips, including approximately 300000 SNPs for the 275 available patient samples. All GWAS quality control and analyses were conducted using PLINK software v 1.07 [9]. After quality control, 273 individuals remained, including 87 African Americans (all 8 available patients with treatment failure vs 79 controls), 40 of whom were receiving EFV regimens (6 with treatment relapse vs 34 controls).
Statistical Analysis
Based on known information about the CYP2B6 EFV slow- metabolizing alleles, analyses were restricted to individuals with phenotype and genotype information who self-reported as African American. After excluding 1 self-reported African American and Hispanic individual (homozygous reference for both CYP2B6 SNPs) and the 1 nonresponder (reported as nonadherent by the site investigator), there were 85 available African American individuals for rs28399499 and 84 for rs3745274.
To determine whether African American patients with the CYP2B6 slow metabolizer allele(s) were at increased risk for HCV relapse, each CYP2B6 SNP (rs3745274 and rs28399499) was tested via Fisher exact test, both independently and in combination. Dominant (homozygous or heterozygous variant vs homozygous reference) and recessive (homozygous variant vs heterozygous or homozygous reference) models were tested, with 2-sided P values and a significance cutoff of P < .05. Analyses were conducted for (1) all African Americans (maximum, n = 85, including 7 with treatment relapse) and (2) African Americans receiving EFV only (n = 40, including 6 with treatment relapse).
RESULTS
For the SNP most associated with increased plasma EFV, rs3745274 (516G → T), there was no association with HCV relapse among African Americans receiving an EFV-containing ARV regimen under either dominant (P = .40) or recessive (P = .58) models (Figure 1A). The observed MAF in this cohort was 44% for African Americans overall and 42.5% for those receiving EFV, which was slightly higher than publicly reported (African, MAF 37% [8]). Likewise, for the rarer EFV-metabolizing SNP rs28399499 (983T → C), there were no significant associations with HCV relapse among African Americans receiving EFV, under a dominant model (P > .99; Figure 1B).
When we investigated the combined effect of both CYP2B6 SNPs by comparing normal EFV metabolizers (homozygous reference for both SNPs) with intermediate or slow metabolizers (heterozygous or homozygous variant at one or both SNPs) there was no significant effect of CYP2B6 genotype on HCV relapse among African Americans receiving EFV (P > .99). A logistic regression model allowing for 3 EFV metabolizer levels (normal metabolizers, homozygous reference for both SNPs; intermediate, 1 variant allele; slow, ≥2 variant alleles) showed a trend toward increased relapse among slow metabolizers (relapse in 9% of normal, 11% of intermediate, and 30% of slow metabolizers), but the effect of CYP2B6 genotype on HCV relapse still failed to reach statistical significance (P = .20; Figure 1C).
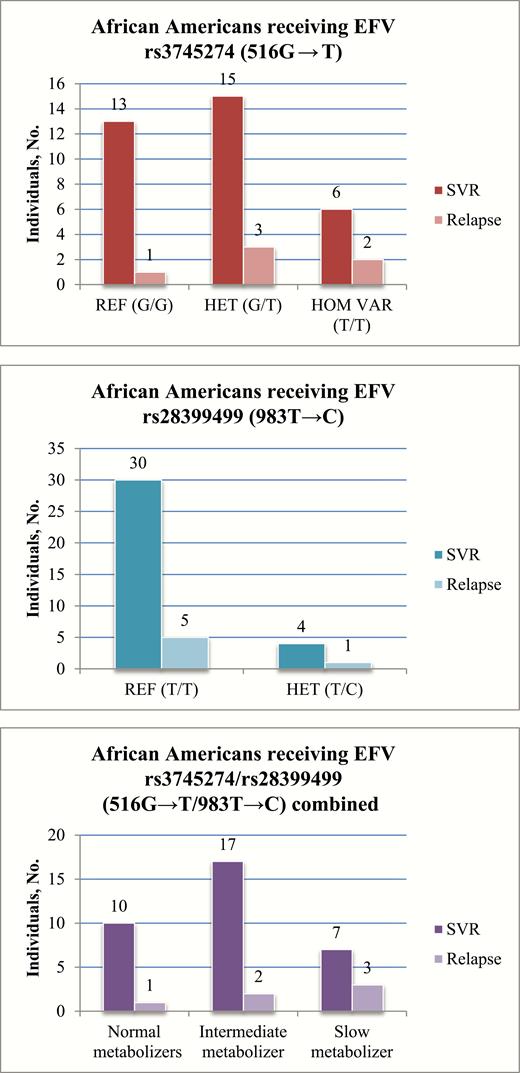
No association was observed between CYP2B6 variant alleles rs3745274 (516G → T) (A) and rs28399499 (983T → C) (B) or the combined rs3745274/rs28399499 (516G → T/983T → C) genotypes (C) and an increased risk of hepatitis C virus (HCV) treatment relapse among human immunodeficiency virus (HIV)–infected African Americans receiving efavirenz (EFV). Normal metabolizers are defined as homozygous reference for both single-nucleotide polymorphisms (SNPs); intermediate metabolizers, 1 variant allele between both SNPs; and slow metabolizers, ≥2 variant alleles between both SNPs.Abbreviations: EFV, efavirenz; HCV, hepatitis C virus; HET, heterozygous for the variant allele; HIV, human immunodeficiency virus; HOM VAR, homozygous for the variant allele (none observed for rs28399499); REF, homozygous for the reference allele; SNPs, single-nucleotide polymorphisms; SVR, sustained virologic response.
Overall, there were no significant associations between CYP2B6 variant alleles rs3745274 (516G → T) and rs28399499 (983T → C) and an increased risk of HCV treatment relapse in African Americans treated with EFV. This was true under dominant and recessive models, and whether the variants were analyzed independently or combined. When the analyses were broadened to risk of HCV treatment relapse among all African Americans in the study, regardless of HIV ARV regimen, the results were similarly nonsignificant (rs3745274, P = .43–.60; rs28399499, P > .99; combined, P = .23 to >.99; data not shown). A subsequent GWAS also failed to detect any common genetic variants significantly associated with HCV treatment failure, though the GWAS was severely underpowered owing to the extremely limited sample size (seeSupplementary Figures S1 and S2).
DISCUSSION
The current report represents a hypothesis-driven investigation, for which there was strong biologic plausibility, of genetic variation affecting LDV/SOF treatment outcome in HIV/HCV-coinfected patients undergoing concurrent ARV therapy. Known EFV slow metabolizer SNPs in CYP2B6 [5] were studied in detail in the available African American subgroup of the ION-4 cohort [1], which included 7 of the 10 HCV treatment relapse events. However, only 40 individuals were both African American and receiving an EFV-containing ARV treatment regimen (including 6 patients with treatment relapse), significantly limiting the power of the study.
Among African American patients in ION-4 receiving concomitant EFV, HCV relapse was not explained by CYP2B6 slow metabolizer genotypes. This was unexpected given that these SNPs increased circulating EFV levels among African Americans in prior studies [4–6] and that a 30% decrease in LDV levels in the presence of EFV was observed in a small phase 1 study in healthy volunteers [7]. However, more recent data from the phase 3 ION-4 trial showed that LDV levels did not differ significantly between HIV/HCV-coinfected patients receiving EFV and those receiving other HIV regimens [10]. Furthermore, there is no evidence of LDV or SOF metabolism by CYP2B6 or CYP3A, the 2 primary EFV metabolizing enzymes [4]. Whereas the extremely small number of individuals experiencing relapse makes it challenging to draw definitive conclusions regarding potential genetic associations, we did not observe evidence of EFV-metabolizing CYP2B6 SNPs influencing LDV/SOF treatment relapse in the current study.
Future research will require larger sample sizes, which may also clarify whether there is a racial/ethnic component to HCV relapse among HIV/HCV-coinfected individuals receiving concurrent ARV therapy. Additional targeted analyses of drug metabolizing genes may be possible, especially as more is understood about the mechanism of LDV metabolism. However, GWAS targeted analyses of annotated drug metabolizing genes could not reach statistical significance at this sample size. Future studies could also investigate the contribution of rare genetic variation, which can play a large role in complex diseases [11, 12].
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Financial support. This work was supported by Gilead Sciences.
Acknowledgments. We acknowledge John McHutchison, MD (Gilead Sciences) for supporting this work and publication.
Potential conflicts of interest. L. M. S. is an employee of and owns stock in Gilead Sciences. M. S. has served as a paid consultant for AbbVie, Gilead, Janssen, and Merck, as a member of a Data Safety Monitoring Board for Gilead (funds paid to Johns Hopkins University), and as a principal investigator for clinical research for AbbVie, Gilead, and Merck (funds paid to Johns Hopkins University); he was also partially supported by a midcareer mentoring award from the National Institutes of Health/National Institute on Drug Abuse (award K24DA034621). D. B. G. has received research funding from Janssen, AstraZeneca, Gilead, Biogen, and UCB; is a consultant for AstraZeneca; has an advisor role for Apostle; holds stock in Pairnomix and Praxis; and holds a patent for IL28B findings. S. N. has received research funds from Gilead, Janssen, Bristol-Myers Squibb, AbbVie, Merck, Vertex Pharmaceuticals, and Tacere and has received personal fees from the Infectious Diseases Society of America; her research institution (Duke University) has also received funding from Eviral Hep, nonfinancial support from the International Antiviral Society–USA, and revenue from Platform Q Health and Practice Point Commons. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References


{kind=link}