






Abstract
Listeriamonocytogenes ( Lm ) causes severe foodborne illness (listeriosis). Previous molecular subtyping methods, such as pulsed-field gel electrophoresis (PFGE), were critical in detecting outbreaks that led to food safety improvements and declining incidence, but PFGE provides limited genetic resolution. A multiagency collaboration began performing real-time, whole-genome sequencing (WGS) on all US Lm isolates from patients, food, and the environment in September 2013, posting sequencing data into a public repository. Compared with the year before the project began, WGS, combined with epidemiologic and product trace-back data, detected more listeriosis clusters and solved more outbreaks (2 outbreaks in pre-WGS year, 5 in WGS year 1, and 9 in year 2). Whole-genome multilocus sequence typing and single nucleotide polymorphism analyses provided equivalent phylogenetic relationships relevant to investigations; results were most useful when interpreted in context of epidemiological data. WGS has transformed listeriosis outbreak surveillance and is being implemented for other foodborne pathogens.
Invasive Listeria monocytogenes ( Lm ) infection (listeriosis) is a rare but serious cause of foodborne illness in the United States, causing an estimated 19% of US deaths from foodborne diseases and costing society an estimated $2.8 billion annually [ 1 , 2 ]. During the 1980s and 1990s, food safety measures targeting ready-to-eat meat and poultry products helped reduce the incidence of listeriosis by >50% [ 3 ]. In 2015, other foods, particularly dairy products and produce items, were identified as the main sources of listeriosis [ 4 ]. Since 2001, listeriosis incidence has changed little despite intensive efforts, remaining above the Healthy People 2020 target of 0.2 cases per 100 000 [ 3 , 5 ].
Although Lm was first recognized as a human pathogen in the 1920s, the first foodborne outbreak of listeriosis was not recognized until the early 1980s [ 6 , 7 ]. This half-century gap between discovery of the pathogen and discovery of its transmission route underscores the difficulty in solving listeriosis outbreaks, an area once known as the “graveyard of epidemiology” [ 8 ]. Listeriosis outbreaks have historically been difficult to solve because they are often small, incubation periods are frequently long [ 9 ], and patients are typically severely ill or deceased, thus limiting statistical power and completeness of food histories.
Only a small proportion of listeriosis cases are linked to recognized outbreaks (ie, 2 or more cases linked to a common food) [ 10 ], but outbreak investigations are important because they provide critical information about food sources of listeriosis and identify food safety gaps. Before nationwide molecular subtyping, US investigators generally detected only large or localized outbreaks. From 1983 (the first recognized US foodborne outbreak of listeriosis) through 1997, 5 listeriosis outbreaks with an identified food source were reported (mean, 0.3 outbreaks/year) with a median of 54 cases per outbreak [ 3 ]. In 1998, PulseNet, a national network of local, state, and federal public health laboratories coordinated by the Centers for Disease Control and Prevention (CDC), began subtyping Lm isolates with pulsed-field gel electrophoresis (PFGE) [ 11 ]. During the next 6 years, investigators solved >5-fold more outbreaks (mean, 2.3 per year); outbreaks were notably smaller (median, 11 cases/outbreak) [ 3 ]. Molecular subtyping has proven critical for identifying clusters that warrant further investigation, but epidemiology is needed to identify or confirm the outbreak food source. (A cluster is a group of isolates linked by molecular subtyping, a group of illness linked by epidemiology, or both. The cluster may constitute an outbreak, but a source, eg, food, has not yet been determined). To enhance collection of epidemiologic information, in 2004 epidemiologists at CDC started the Listeria Initiative (LI) [ 12 ], modeled after a similar program in France. Participating state and local health departments interview all listeriosis patients (or surrogates) as soon as possible after diagnosis using a standardized questionnaire asking about >40 different food exposures. When molecular subtyping detects a listeriosis cluster, epidemiologists use LI data to compare food exposures among patients in the cluster with exposures of patients with non-outbreak-associated (sporadic) listeriosis to identify a possible food vehicle. During 2004–2013, the first decade of LI use, investigators solved more outbreaks (mean, 3.5 per year) with fewer cases per outbreak (median, 5.5) than in previous years.
Although PFGE has proved remarkably useful in detecting listeriosis clusters, it lacks the discriminatory capacity and phylogenetic basis of more advanced methods [ 13 ]. Genome insertions, deletions, rearrangement, and point mutations at a restriction enzyme site can cause Lm isolates that are highly related genetically to appear different by PFGE. Similarly, isolates that are not highly related may appear indistinguishable by PFGE. Both scenarios can lead to misclassification when PFGE is used to define an outbreak-related case, which hampers cluster detection and epidemiologic investigations.
WGS-ENHANCED L. MONOCYTOGENES SURVEILLANCE
To enhance listeriosis surveillance and control by providing improved understanding of phylogenetic relationships among Lm isolates, in September 2013, CDC, the US Food and Drug Administration (FDA), the US Department of Agriculture's Food Safety and Inspection Service (USDA-FSIS), the National Institute for Biotechnology Information (NCBI), and local, state, and international partners began a pilot project to prospectively perform whole-genome sequencing (WGS) on all available Lm isolates collected from patients, food, and food processing environments in the United States. Sequencing-based subtyping methods have previously been used successfully in investigation of outbreaks detected by other methods (eg, PFGE) [ 14–21 ], but never before for real-time foodborne disease surveillance. Lm was the ideal foodborne pathogen for such a pilot because of its relatively small and simple genome, the severity of the disease it causes, strong epidemiologic surveillance, and frequent testing for contamination in ready-to-eat foods. At the end of the pilot, the partner agencies incorporated WGS into standard national listeriosis surveillance.
Beginning in 2012, FDA created a network of GenomeTrakr laboratories, which now consists of 30 sites (27 domestic, 3 international) that sequence Lm and other foodborne pathogen isolates from food and environmental sources. State regulatory laboratories also sequenced food and environmental isolates and uploaded data into the GenomeTrakr database. The WGS Lm pilot began in September 2013 when CDC started sequencing all Lm isolates from patients. Over time, state and local public health laboratories developed the capacity to sequence isolates locally. As of March 2016, 20 of these laboratories were sequencing all Lm patient isolates and another 7 were in preparation. USDA-FSIS sequences all Lm isolates obtained from sampling of meat and poultry products and FSIS-regulated food processing environments. PFGE is performed in parallel with WGS.
All participating agencies submit raw sequence data that meet minimum quality thresholds and metadata (eg, date, location, submitting laboratory, sequencing parameters, source of isolates) to publicly accessible NCBI data archives (BioSample and Sequence Read Archive) under a single BioProject [ 22 ]. Metadata for patient isolates are limited to protect patient confidentiality. NCBI continually generates and shares phylogenies constructed from all Lm with publicly available genomes [ 23 ].
CDC collaborated with international partners to identify the core genome and create a whole-genome multilocus sequence typing (wgMLST) scheme for Lm [ 24 ]. The MLST analysis method is a gene-centric process whereby allelic differences are counted at the gene level [ 25 ]. For Lm , there are 7 gene fragments in the basic housekeeping MLST scheme [ 26 ] and 4804 in wgMLST, in which each gene is found in nearly all Lm genomes. These MLST schemes have been built into the existing software used to manage PulseNet data (BioNumerics 7.5, Applied Maths) [ 24 ]. The software determines whether the nucleotide sequence among isolates is identical or different at each locus or gene. For example, when comparing 2 isolates, any sequence differences (eg, 1 or >100 single-nucleotide polymorphisms [SNPs]) at a single locus count as a single allelic difference; if the 2 isolates have identical sequences at that locus, no allelic difference is counted. This method allows users to quickly and efficiently infer phylogenetic trees with relatively little bioinformatics expertise, allowing state and local public health laboratories to independently perform wgMLST analyses and query the national database for highly related isolates.
The FDA, CDC, and USDA-FSIS also analyze WGS data using free, open-source SNP-based approaches, which create phylogenetic trees based on nucleotide differences among genomes under investigation that are mapped for comparison to a reference genome [ 27 , 28 ]. For identifying high-quality SNP (hqSNP) differences among genomes, the SNP positions are filtered for sequence depth and quality. These hqSNP phylogenies provide a high level of detail, but the analysis can be computationally intensive for large numbers of isolates and typically requires a skilled bioinformatician to do the analysis.
IMPACT OF L. MONOCYTOGENES WGS PROJECT ON OUTBREAK INVESTIGATIONS
WGS has transformed listeriosis outbreak surveillance and response. Compared with the year before comprehensive WGS began (September 2012–August 2013), more clusters were detected in each year following introduction of WGS (14 in the pre-WGS year, 19 in year 1, and 21 in year 2) (Figure 1 ). Improvement in numbers of clusters detected sooner or only by WGS compared with PFGE and a marked reduction in median cluster size were also observed. Similarly, more outbreaks were solved following implementation of WGS (2 outbreaks in the pre-WGS year, 5 outbreaks in year 1, and 9 outbreaks in year 2), linking more cases to food sources.
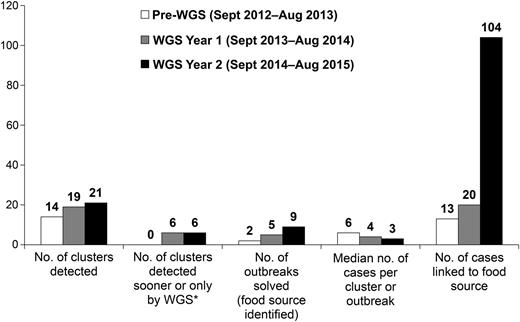
Listeriosis clusters detected and outbreaks solved before and after implementation of real-time whole-genome sequencing (WGS) of Listeria monocytogenes isolates from patients, food, and the environment, United States, September 2012–August 2015. *Cluster detection sooner or only by WGS, as compared with pulsed-field gel electrophoresis.
Based on experiences to date, WGS has improved listeriosis outbreak investigations in at least 6 key ways; an example is provided for each.
Delineated clusters with diverse PFGE patterns: A multistate outbreak linked to a major commercial producer of ice cream was initially detected at a single hospital in Kansas [ 29 ]. During a 13-month period, 5 patients developed listeriosis following hospitalization for unrelated reasons, which prompted an investigation. The first 3 patients had isolates with different PFGE patterns, making it appear less likely that the infections were related. When the fourth patient was found to have a PFGE pattern indistinguishable from one of the others, an epidemiologist from the state health department contacted CDC. Four isolates were highly related by wgMLST (≤18 allele differences). CDC had recently learned that product samples from ice cream producer A had yielded Lm with 7 different PFGE patterns; these isolates were found to be highly related by wgMLST to isolates from patients (≤28 allele differences). Epidemiological information supported this connection: All 4 patients with available information consumed milkshakes made with ice cream from producer A.
Determined the source of “cold cases”: In December 2013, PulseNet identified a cluster of 4 listeriosis cases in a single state with indistinguishable PFGE patterns. Local, state, and CDC epidemiologists investigated commonalities among patients and found that all were of Middle Eastern descent. At the time, no common food was identified because of limited exposure information. In August 2015, PulseNet identified a multistate cluster of 20 Lm isolates that were highly related by wgMLST (≤25 alleles). The patients' isolates had 5 different PFGE patterns and specimen collection dates ranging from April 2013 to August 2015, including the 4 cases from the previous investigation. With the additional exposure information available for this 5-fold larger number of cases, investigators traced the outbreak's source to a single cheese producer [ 30 ]. WGS helped link the source of the previously unsolved cluster from the 2013 investigation to the same cheese producer. Retrospective sequencing showed that cheese from this producer was the source of additional unsolved cases dating back to 2010. WGS allows investigators to link cases that occurred over a longer time period than previously used (ie, several years vs several months), allowing identification of low-level product contamination and the effects of preventive controls.
Demonstrated that certain PFGE-defined clusters did not consist of highly related isolates: In October 2013, PulseNet identified a multistate cluster of Lm isolates by PFGE. Despite considerable efforts, including time spent investigating a possible link to deli meats, investigators could not identify a common source. In January 2014, after hqSNP analyses showed that patient isolates were not highly related (most isolates differed by >30 hqSNPs, also by >30 alleles) (Figure 2 ), this resource-intensive investigation ended. Averting such PFGE “pseudo-clusters” saves limited public health resources at the federal, state, and local levels.
Refined outbreak case definitions: In July 2014, PulseNet detected a PFGE cluster of Lm isolates from 8 patients in 5 states. Limited epidemiologic information was available to implicate a source food. In August, hqSNP analysis showed that 4 of these isolates were highly related (≤3 hqSNPs, ≤6 alleles), but 4 were not (differed by >60 hqSNPs, >50 alleles) and were excluded from the investigation. The remaining 4 patients resided within a single metropolitan area, suggesting a locally produced food as the likely source. In September, FDA uploaded to PulseNet indistinguishable isolates from mung bean sprouts and environmental samples from a sprout-producing facility located in that metropolitan area. The 2 patients with available information reported eating food containing mung bean sprouts. WGS later showed that the food and environmental isolates were highly related to the patient isolates (≤3 hqSNPs, ≤6 alleles), prompting a public announcement about the outbreak that would not have been possible with PFGE and limited epidemiologic data alone [ 31 ]. An important caveat to excluding cases by WGS is that outbreaks can be polyclonal. For example, in an outbreak linked to caramel apples [ 32 ], 2 distinct groups of highly related isolates (both from patients and apples) differed from one another at >50% of alleles.
Link sporadic illnesses to contaminated food: Before WGS, linking a sporadic case of listeriosis to a specific food was rarely possible without isolating Lm from a contaminated food in the patient's home. In March 2014, a firm recalled certain types of prepackaged lettuce after a regulatory agency found Lm during routine testing. No illnesses were known to be linked to the recall. The isolate from lettuce had the sixth most common PFGE pattern (of >2500) for Lm in the United States, so with PFGE alone it would have been difficult to establish a link to contemporaneous patient isolates in PulseNet. However, WGS showed that an isolate from a patient in Ohio and the lettuce isolate were highly related [ 33 ], differing by only 4 alleles and by 5 hqSNPs. This patient reported regularly consuming multiple types of prepackaged lettuce, including the recalled brand. No other patient isolates in PulseNet were closely related to these 2 isolates. The WGS and epidemiologic data provided strong circumstantial evidence of a link between a single illness and the recalled food.
Confirm outbreaks following product testing: In July 2014, a firm recalled peaches, nectarines, and other stone fruit after company testing of fruit detected Lm . No illnesses were known to be linked to the recall. In August, 3 Lm isolates from samples of recalled stone fruit were uploaded to PulseNet and found to have indistinguishable PFGE patterns; 4 patient isolates with specimen collection dates in May–August 2014 also shared this pattern. Two of these 4 patient isolates were highly related to the stone fruit isolates, each differing by <10 alleles and <10 hqSNPs [ 34 ]. Both patients reported eating stone fruit, and shopper card data confirmed that the stone fruit eaten by one had been recalled. Interview data showed that 1 patient with an isolate not highly related by wgMLST to stone fruit isolates did not eat these fruit. The other patient could not be reached for interview. Thus, WGS and epidemiologic data confirmed that 2 illnesses could be linked to stone fruit.
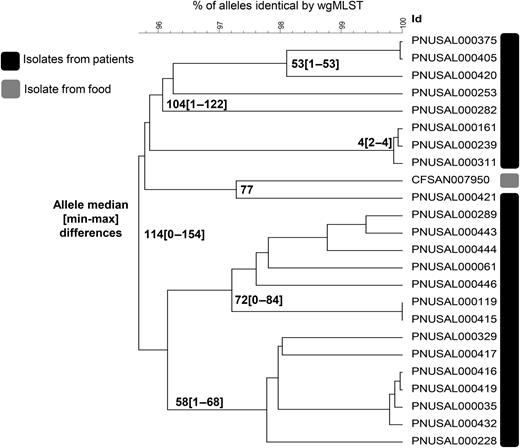
Phylogenetic tree showing whole-genome multilocus sequence typing (wgMLST) allele differences among Listeria monocytogenes isolates from patients and food that were indistinguishable by pulsed-field gel electrophoresis (PFGE) and were identified as a disease cluster for investigation, United States, 2013 (N = 24). Most isolates were not highly related, with >30 allele differences between them, indicating this PFGE-defined cluster was a “pseudo-cluster” by whole-genome sequencing.
In the longer term, WGS can improve our understanding of the ecology of Lm contamination in the food production environment. In 3 small outbreaks (≤6 cases each) linked to soft cheese (2 outbreaks) and sprouts (1 outbreak) [ 31 , 35 , 36 ], relatively few allelic differences (≤12) existed among all patient, food, and environmental isolates, possibly because all involved small production environments. Strains harbored within some facilities appear to change little over time. For example, in 3 outbreak investigations during 2013–2015 linked to soft cheese [ 30 , 35 , 36 ], food and environmental isolates collected from the same facility 3–5 years apart were found to be highly related by WGS (≤13 alleles). By comparison, a more diverse phylogenetic range of isolates has been observed in outbreaks linked to large companies. In the outbreak linked to large ice cream producer A [ 29 ], isolates from one production facility differed by up to 30 alleles (excluding 4 food isolates that were >200 alleles from others), and isolates from a second differed by up to 32 alleles (Table 1 ). Similarly, in the outbreak linked to stone fruit [ 34 ], there were up to 43 allele differences among isolates. Factors that may influence the diversity of isolates within an outbreak include the source of food contamination (ie, whether on the farm or during transport or processing), the type of food, the number and manner of Lm introductions into a food processing environment, the size of a food processing facility, and the environmental conditions and duration in which Lm survives and grows before contaminating food.
Whole-Genome Sequencing Analyses by High-Quality Single-Nucleotide Polymorphism and Whole-Genome Multilocus Sequence Typing of Listeria monocytogenes Isolates From Patients, Foods, and the Environment in the 8 Multistate Outbreaks of Listeriosis Solved in the United States During September 2012–August 2015
| WGS Year a | No. of Cases | Implicated Food | No. of Food and Environmental Isolates | Maximum No. of Differences Among Outbreak-Related Isolates (Median) | Reference | |
|---|---|---|---|---|---|---|
| hqSNPs | Alleles by wgMLST | |||||
| Pre-WGS | 6 | Soft cheese (French-style) | 2 | 8 (1) | 5 (1.5) | [ 36 ] |
| Year 1 | 8 | Soft cheese (Latin-style) | 37 | 18 (7.5) | 19 (5) | [ 37 ] |
| Year 1 | 5 | Mung bean sprouts | 36 | 25 (6) | 14 (5) | [ 31 ] |
| Year 1 | 2 | Stone fruit | 46 | 44 (2) | 43 (10) | [ 34 ] |
| Year 2 | 9 | Soft cheese (Latin-style) | 7 | 21 (16) | 22 (14) | [ 38 ] |
| Clade 1: 18 (10) | Clade 1: 11 (3) | |||||
| Year 2 | 35 | Caramel apples | 32 | Clade 2: 10 (4) b | Clade 2: 8 (3) b | [ 32 ] |
| Year 2 | 4 | Ice cream (from production facility 1) | 144 | 31 (11) c | 30 (15) c | [ 29 ] |
| Year 2 | 5 | Ice cream (from production facility 2) | 51 | 67 (17) d | 32 (15) d | [ 29 ] |
| Year 2 | 34 | Soft cheese (Middle Eastern and European-style) | 9 | 38 (16) | 26 (13) | [ 30 ] |
| WGS Year a | No. of Cases | Implicated Food | No. of Food and Environmental Isolates | Maximum No. of Differences Among Outbreak-Related Isolates (Median) | Reference | |
|---|---|---|---|---|---|---|
| hqSNPs | Alleles by wgMLST | |||||
| Pre-WGS | 6 | Soft cheese (French-style) | 2 | 8 (1) | 5 (1.5) | [ 36 ] |
| Year 1 | 8 | Soft cheese (Latin-style) | 37 | 18 (7.5) | 19 (5) | [ 37 ] |
| Year 1 | 5 | Mung bean sprouts | 36 | 25 (6) | 14 (5) | [ 31 ] |
| Year 1 | 2 | Stone fruit | 46 | 44 (2) | 43 (10) | [ 34 ] |
| Year 2 | 9 | Soft cheese (Latin-style) | 7 | 21 (16) | 22 (14) | [ 38 ] |
| Clade 1: 18 (10) | Clade 1: 11 (3) | |||||
| Year 2 | 35 | Caramel apples | 32 | Clade 2: 10 (4) b | Clade 2: 8 (3) b | [ 32 ] |
| Year 2 | 4 | Ice cream (from production facility 1) | 144 | 31 (11) c | 30 (15) c | [ 29 ] |
| Year 2 | 5 | Ice cream (from production facility 2) | 51 | 67 (17) d | 32 (15) d | [ 29 ] |
| Year 2 | 34 | Soft cheese (Middle Eastern and European-style) | 9 | 38 (16) | 26 (13) | [ 30 ] |
Abbreviations: hqSNPs, high-quality single-nucleotide polymorphisms; wgMLST, whole-genome multilocus sequence typing; WGS, whole-genome sequencing.
a Pre-WGS year is September 2012–August 2013, Year 1 is September 2013–August 2014, and Year 2 is September 2014–August 2015.
b Clades were median of 7494 hqSNPs and 1635 alleles different.
c Four additional food isolates were up to 277 hqSNPs and 239 alleles from others from facility 1.
d Up to 387 hqSNPs and 265 alleles between isolates from facilities 1 and 2 of producer A. Cases linked to the 2 facilities are considered part of the same outbreak.
Whole-Genome Sequencing Analyses by High-Quality Single-Nucleotide Polymorphism and Whole-Genome Multilocus Sequence Typing of Listeria monocytogenes Isolates From Patients, Foods, and the Environment in the 8 Multistate Outbreaks of Listeriosis Solved in the United States During September 2012–August 2015
| WGS Year a | No. of Cases | Implicated Food | No. of Food and Environmental Isolates | Maximum No. of Differences Among Outbreak-Related Isolates (Median) | Reference | |
|---|---|---|---|---|---|---|
| hqSNPs | Alleles by wgMLST | |||||
| Pre-WGS | 6 | Soft cheese (French-style) | 2 | 8 (1) | 5 (1.5) | [ 36 ] |
| Year 1 | 8 | Soft cheese (Latin-style) | 37 | 18 (7.5) | 19 (5) | [ 37 ] |
| Year 1 | 5 | Mung bean sprouts | 36 | 25 (6) | 14 (5) | [ 31 ] |
| Year 1 | 2 | Stone fruit | 46 | 44 (2) | 43 (10) | [ 34 ] |
| Year 2 | 9 | Soft cheese (Latin-style) | 7 | 21 (16) | 22 (14) | [ 38 ] |
| Clade 1: 18 (10) | Clade 1: 11 (3) | |||||
| Year 2 | 35 | Caramel apples | 32 | Clade 2: 10 (4) b | Clade 2: 8 (3) b | [ 32 ] |
| Year 2 | 4 | Ice cream (from production facility 1) | 144 | 31 (11) c | 30 (15) c | [ 29 ] |
| Year 2 | 5 | Ice cream (from production facility 2) | 51 | 67 (17) d | 32 (15) d | [ 29 ] |
| Year 2 | 34 | Soft cheese (Middle Eastern and European-style) | 9 | 38 (16) | 26 (13) | [ 30 ] |
| WGS Year a | No. of Cases | Implicated Food | No. of Food and Environmental Isolates | Maximum No. of Differences Among Outbreak-Related Isolates (Median) | Reference | |
|---|---|---|---|---|---|---|
| hqSNPs | Alleles by wgMLST | |||||
| Pre-WGS | 6 | Soft cheese (French-style) | 2 | 8 (1) | 5 (1.5) | [ 36 ] |
| Year 1 | 8 | Soft cheese (Latin-style) | 37 | 18 (7.5) | 19 (5) | [ 37 ] |
| Year 1 | 5 | Mung bean sprouts | 36 | 25 (6) | 14 (5) | [ 31 ] |
| Year 1 | 2 | Stone fruit | 46 | 44 (2) | 43 (10) | [ 34 ] |
| Year 2 | 9 | Soft cheese (Latin-style) | 7 | 21 (16) | 22 (14) | [ 38 ] |
| Clade 1: 18 (10) | Clade 1: 11 (3) | |||||
| Year 2 | 35 | Caramel apples | 32 | Clade 2: 10 (4) b | Clade 2: 8 (3) b | [ 32 ] |
| Year 2 | 4 | Ice cream (from production facility 1) | 144 | 31 (11) c | 30 (15) c | [ 29 ] |
| Year 2 | 5 | Ice cream (from production facility 2) | 51 | 67 (17) d | 32 (15) d | [ 29 ] |
| Year 2 | 34 | Soft cheese (Middle Eastern and European-style) | 9 | 38 (16) | 26 (13) | [ 30 ] |
Abbreviations: hqSNPs, high-quality single-nucleotide polymorphisms; wgMLST, whole-genome multilocus sequence typing; WGS, whole-genome sequencing.
a Pre-WGS year is September 2012–August 2013, Year 1 is September 2013–August 2014, and Year 2 is September 2014–August 2015.
b Clades were median of 7494 hqSNPs and 1635 alleles different.
c Four additional food isolates were up to 277 hqSNPs and 239 alleles from others from facility 1.
d Up to 387 hqSNPs and 265 alleles between isolates from facilities 1 and 2 of producer A. Cases linked to the 2 facilities are considered part of the same outbreak.
In our experience investigating listeriosis clusters with wgMLST (ie, allele) and hqSNP analyses, the 2 methods equally distinguish isolates belonging to an outbreak from sporadic cases with high epidemiological concordance [ 24 , 39 ]. The numbers of allele and hqSNP differences between isolates are typically similar when there are <100 differences (alleles or hqSNPs) between isolates. Theoretically, this concordance arises because point mutations typically do not occur twice in the same gene in closely related isolates.
European investigators have reported that a cutoff of ≤10 alleles by core genome MLST could distinguish outbreak-related isolates from unrelated ones in 2 outbreaks [ 40 ]. Our experience investigating a wider variety of clusters and outbreaks suggests that that a single cutoff cannot consistently predict whether isolates will be epidemiologically related. Isolates with <10 wgMLST allele or hqSNP differences were often epidemiologically linked, whereas those in the 10–30 range were frequently linked, and isolates with >30 differences were occasionally linked. WGS results should not be interpreted in isolation, and other contextual information (ie, epidemiologic or product trace-back data) is needed to confirm the source.
CONCLUSIONS AND FUTURE DIRECTIONS
In the context of robust epidemiologic data collection and regulatory food testing, the increased discriminatory capacity of WGS has helped strengthen links among Lm isolates from food, the environment, and patients to detect more listeriosis clusters, halt pseudo-cluster investigations, focus epidemiologic investigations, and ultimately, solve more outbreaks. In several instances, the high resolution of WGS allowed investigators and regulatory agencies to take action at a lower level of epidemiological evidence, a key advantage for the relatively small outbreaks typical for listeriosis. By helping to solve more outbreaks, WGS has increased public awareness of listeriosis, focused increased attention on contamination that food companies can prevent, and improved understanding of Lm in food. WGS, by helping to uncover food safety gaps through outbreak investigations, complements the ongoing implementation of the Food Safety Modernization Act, which aims to improve measures to prevent foodborne illness [ 41 ]. These interventions should lead to renewed reductions in Lm -related illnesses and deaths. Based on the success of WGS-enhanced listeriosis surveillance, CDC and partner agencies are expanding the technology to US surveillance for other pathogens transmitted commonly by food, including Shiga toxin–producing Escherichia coli , Campylobacter , and Salmonella .
The Lm wgMLST analysis tool, previously only available at CDC, will soon be available to all US PulseNet member laboratories. Because standardization is essential for public health surveillance, CDC, FDA, and USDA-FSIS continue to work with academic and international partners to set sequence quality benchmarks and nomenclature schemes. As WGS becomes more integrated into foodborne outbreak detection, public health officials at all levels will need to learn core concepts about the technology and its interpretation, particularly as many will need to describe the implications of these analyses to industry representatives, policymakers, and the media.
Notes
Acknowledgments. Kristin G. Holt, John Johnston, Frankie Beacorn, Cathy Pentz, and Todd Lauze of US Department of Agriculture's Food Safety and Inspection Service (USDA-FSIS) also contributed to the Lm whole-genome sequencing (WGS) pilot project. We thank our international collaborators, including Sylvain Brisse, Marc Lecuit, Alexander LeClercq, Alexandra Moura, and colleagues at Institut Pasteur, as well as colleagues at the Public Health Agency of Canada. We acknowledge the critical work of state and local health departments, laboratories, and regulatory agencies in foodborne disease outbreak detection and investigation and in rapidly implementing WGS in their public health work.
Disclaimer. The findings and conclusions in this paper are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention (CDC), the Food and Drug Administration (FDA), FSIS, or the National Institutes of Health (NIH).
Financial support. This work was supported by CDC, including CDC's Advanced Molecular Detection Initiative and Cooperative Agreement number U60HM000803; FDA; USDA-FSIS; National Institute for Biotechnology Information; and the Intramural Research Program of the NIH, National Library of Medicine. H. P. is an employee of Applied Maths.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References


{kind=link}
{kind=link}