





Abstract
Systemic sclerosis (SSc) is considered a rare autoimmune disease in which there are alterations of both the innate and adaptive immune response resulting in the production of autoantibodies. Abnormalities of the immune system compromise the normal function of blood vessels leading to a vasculopathy manifested by Raynaud’s phenomenon, an early sign of SSc . As a consequence of this reactive picture, the disease can evolve leading to tissue fibrosis. Several SSc-specific autoantibodies are currently known and are associated with specific clinical manifestations and prognosis. Although the pathogenetic role of these autoantibodies is still unclear, their production by B cells and plasma cells suggests the importance of these cells in the development of SSc. This review narratively examines B-cell dysfunctions and their role in the pathogenesis of SSc and discusses B-cell-targeted therapies currently used or potentially useful for the management of end-organ complications.
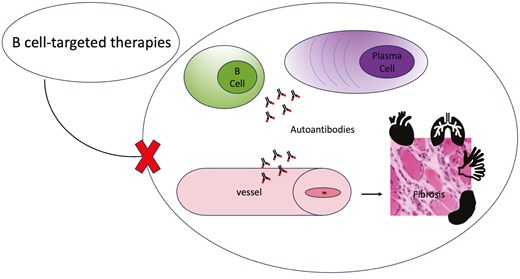
Introduction
Systemic sclerosis (SSc) is a rare autoimmune rheumatic disease [1] characterized by endothelial cells dysfunction and microvascular changes and a complex modification of the immune response involving innate and adaptive immunity with production of autoantibodies [2]. The interplay between the immune system and the vessels leads to vasculopathy which is clinically evident with Raynaud’s phenomenon which is the first and early onset sign [3–5]. The immune activation and the vasculopathy eventually contribute to the evolution to fibrosis in the skin and internal organs [6], lungs and gastrointestinal system being the most frequently involved systems [7].
In SSc, different SSc-specific autoantibodies can be detected: anticentromere autoantibodies (ACAs) in limited SSc (lcSSc), anti-topoisomerase I autoantibodies (ATAs) mainly in diffuse SSc (dcSSc) [8], while anti-RNA polymerase III (RNAP3) autoantibodies are associated with renal involvement, gastric antral venous ectasia, and notably, a rapidly progressive presentation and increased risk of cancer [9]. Other autoantibodies have been described, helping to stratify patients with different organ involvements, complications, and prognosis [10].
Even though the pathogenetic role of these disease-specific autoantibodies has not yet been clearly established [11], their strong association with clinical phenotypes and prognosis suggests a pivotal role of autoantibodies producing B-cells and plasma cells in either the onset or the development of SSc [12–14]. Concurrently, various other dysfunctions in distribution, activation, and signalling of B-cell subsets have been described [15–18].
Herein, the literature on B-cell dysfunctions and on their role in SSc pathogenesis are narratively revised. Additionally, an insight into B-cell-targeted therapies currently used or potentially beneficial for managing organ complications is also provided.
Role of B cells in SSc
In the human immune system, B cells have pleiotropic functions, including antibody and cytokine production, antigen presentation to T cells, modulation of dendritic cell function, and lymphoid organogenesis [19]. Thus, B cells and their antibodies are fundamental actors in humoral immunity, in particular enacting the protection of the human body, as part of the adaptive immune system, against an almost unlimited variety of pathogens. Any defect in B-cell development, selection, and function may lead to malignancy, allergy, immunodeficiencies, and autoimmunity [20]. In the context of autoimmune diseases like SSc, aberrant activation and dysregulation of B cells are today considered a central event in the disease pathogenesis [21].
B-cell subsets
B cells can be broadly classified into several subsets based on their function, and their surface markers may be used as therapeutic targets [22].
Transitional B cells are a distinct subset of B cells found in the transitional stage between immature and mature B cells in the bone marrow and during their early circulation in the periphery [23]. These cells are involved in the development and maturation of the B-cell repertoire and the establishment of immune tolerance [24], and they express markers such as CD19, CD24, CD38++, IgM, and low levels of surface IgD [25]. Thanks to their functional properties, they may respond to antigens, despite the fact that they have not yet undergone the extensive selection and maturation processes that mature B cells have experienced. As a result, they exhibit a degree of functional immaturity. It is also important to point out that transitional B cells play a role in immune tolerance by undergoing selection processes that eliminate autoreactive B cells, thereby preventing the development of autoimmune diseases [26]. In fact, it is important to remark that the dysregulation of transitional B-cell development or function may contribute to the breakdown of immune tolerance and the development of autoimmune conditions [19].
Naïve B cells are a cell subset which have not yet encountered an antigen (although they are mature), and for this reason they are considered ‘naïve’ due to the fact that they have not been activated by exposure to a specific antigen. Naïve B cells play a crucial role in the adaptive immune response by providing a reservoir of antigen-specific cells ready to respond to future encounters with pathogens. These cells express surface markers such as CD19, CD20, CD21, CD22, and immunoglobulin receptors, including IgD and IgM, that allow them to recognize and bind to antigens [27]. Naïve B cells have a high degree of antigen sensitivity but lack effector functions until they encounter and are activated by a specific antigen. When a naïve B-cell encounters its specific antigen, matching their BCRs, they are activated undergoing clonal expansion. This activation leads to the proliferation and differentiation of the naïve B cell into plasma cells or memory B cells.
Memory B cells are the specialized subset of B lymphocytes previously exposed to antigens and have undergone activation and differentiation. Unlike naive B cells that have not encountered antigens yet, memory B cells are primed to mount a rapid and robust secondary immune response upon re-exposure to the same antigen. They are a critical component of the adaptive immune system and are fundamental in immunological memory. This subset of cells bears surface markers such as CD19, CD20, CD27 and may express low levels of IgD and IgM compared with naive B cells [28].
However, they typically excess higher levels of isotype-switched immunoglobulins, such as IgG, IgA, or IgE, that are indicative of their antigen-experienced status. A characteristic of memory B bells is longevity: they are long-lived cells, persisting in the body for extended periods, sometimes for a lifetime. Their longevity allows long-term protection against recurrent infections by maintaining immunological memory [29]. Moreover, memory B cells contribute to the diversity of the immune repertoire by retaining the specificity for the antigen encountered during the primary immune response. This diversity ensures that the immune system can recognize and respond effectively to a wide range of pathogens upon re-encounter [30].
Plasma cells are specialized mature B lymphocytes that have undergone differentiation into antibody-secreting cells. Unlike memory B cells, plasma cells do not express typical B-cell surface markers, excluding CD19, that have been downregulated during their terminal differentiation. Instead, plasma cells express a distinct set of surface markers and proteins associated with their function of antibody secretion. Plasma cells are characterized by the co-expression of CD138 and CD38 [31] and are deeply involved in the adaptive immune response by producing large quantities of antibodies targeting-specific antigens. They are derived from activated B cells, a process mediated by various signalling pathways and transcription factors, and they are responsible for the bulk of antibody production during an immune response. These cells are highly specialized for the production and secretion of antibodies of different isotypes (such as IgG, IgA, IgM, IgE, and IgD) through a process called isotype switching [30]. This process allows plasma cells to tailor the antibody response to different types of pathogens and immune functions. Plasma cells have a short lifespan compared to memory B cells. While some plasma cells may persist for a few days to weeks, the majority undergo programmed cell death (apoptosis), after fulfilling their role in antibody production.
However, new plasma cells can be continuously generated from activated B cells as needed to maintain antibody levels during an ongoing immune response [32]. For this reason, plasma cells are essential components of the adaptive immune system, responsible for generating humoral immunity through the production of antibodies to neutralize pathogens, promote their clearance, and contribute to immune regulation and homeostasis.
It is today well known that the dysregulation of memory B cells and plasma cells can lead to the development of an autoimmune condition [33].
The cellular subsets described so far have the function of promoting the immune response but there are also B cells with a regulatory function. In fact, B regulatory cells (Bregs) play a crucial role in regulating immune responses and maintaining immune tolerance. Unlike conventional B cells, which primarily function to produce antibodies, Bregs are involved in suppressing excessive immune responses and preventing autoimmune reactions. This occurs through the production of anti-inflammatory cytokines, of which interleukin-10 (IL-10) is one of the most important. This cytokine can exert a suppressive mechanism on many cells of the immune system, including T cells. In studies of SSc patients and some mouse models, decreased IL-10 production correlates with decreased IL-10-producing Bregs cells. SSc patients with decreased IL-10 production are associated with the presence of ILD, suggesting their importance in controlling the profibrotic environment [33].
Involvement of B cells in SSc
Numerous studies have highlighted the significant implications of B cells in SSc, where they are found in damaged organs [34–36], their subpopulations are dysregulated [19] and show an activated phenotype [37]. In SSc, B cells play a multifaceted pathogenetic role, contributing to various aspects of the disease process including an abnormal accumulation of collagen and other extracellular matrix components, eventually leading to tissue fibrosis [33]. In this fibrosing process, the production of autoantibodies by B cells and plasma cells is considered a relevant event. We know that in SSc the prevalence of anti-nuclear antibodies (ANAs) is very high (90–95%) [13] and for this reason SSc-specific autoantibodies (ATA, ACA, and ARA) have been included in the 2013 American College Rheumatology/European League Against Rheumatism (ACR/EULAR) SSc classification criteria [38]. However, other autoantibodies like anti-U3-RNP antibody (U3RNP) and less frequently anti-Th/To (Th/To), anti-Ku (Ku), anti-PmScl (PmScl), anti Ro52, and anti-U1-RNP (U1RNP) can be detected [39]. The clinical role of autoantibodies is corroborated by the evidence that they are associated with different phenotypes and are important both for stratifying patients based on the risk of developing organ complications and for overall survival [40].
On top of the pivotal role played by autoantibodies, it is important to remark the role that the production of cytokines by B-cell subsets is functionally playing in SSc pathogenesis. Indeed, B cells produce pro-inflammatory cytokines such as interleukin-6 (IL-6), tumour necrosis factor (TNF)-α, transforming growth factor-β, and interleukin-13 (IL-13), thus promoting inflammation and contributing to endothelial dysfunction, recruitment and activation of fibroblasts and immune cells, eventually promoting extracellular matrix synthesis and driving the progression to fibrosis. This process results in tissue damage and the loss of its function anywhere in the body [19, 41].
Furthermore, B cells interact with T cells modulating their responses. In fact, antigen presentation and co-stimulation of B cells to T cells contributes to activate CD4+ T-cell through small amounts of antigen, which is responsible for the development of autoimmune diseases [16]. The aberrant activation and dysregulation of the B-T cell interaction together with the altered frequencies and functional abnormalities of B-cell subsets, may definitively perpetuate an autoimmune response, a defective tolerance mechanism, a breakdown of self-tolerance and a chronic inflammatory process.
In SSc pathogenesis, another pathway independent from antigen-mediated mechanisms, is the B-cell activating factor (BAFF) pathway: BAFF is a member of the TNF superfamily, playing a critical role in the activation, survival, and maturation of B cells, being involved in many disorders such as transplantation/graft-versus-host disease and autoimmune diseases [42].
These data indicate that understanding the mechanisms underlying B-cell-mediated immune dysregulation in SSc is critical for the development of selective targeted therapies that may block or slow the progression of the disease pathogenesis.
Autoreactive B cells play a central role in autoimmunity thanks to their production of autoantibodies, immune complexes, activation of effector functions, promoting a persistent inflammatory state of tissues with consequent damage [43]. Physiologically, during B-cell development, autoreactive B cells are inactivated or eliminated to prevent autoimmune responses. However, in autoimmune conditions, these tolerance mechanisms can fail or be ignored, leading to the survival and activation of autoreactive B cells [44]. Furthermore, autoreactive B cells can become dysregulated, leading to increased proliferation, survival, and differentiation into antibody-secreting plasma cells [45]. Although autoreactive B-cell responses to nuclear proteins are related to the pathogenesis of SSc, their pathogenetic role still remains elusive [46].
In conclusion, despite some good results shown in vivo, overall, the B-cell-depleting monoclonal strategy is still under investigation in SSc, mainly because autoreactive B cells may persist and still remain endowed in the tissues of internal inflamed organs as well as in lymph nodes.
B-cell-targeted therapies
The identification of the B-cell-dependent mechanisms that may influence either the development or the progression of SSc is instrumental to provide the evidence to create B-cell depleting therapies [47, 48]. Inhibitors of CD20, CD19, BAFF and novel strategies such as CAR-T cell therapy represent today very promising approaches to achieve an early and sustained disease remission. The key mechanisms of the anti CD20 and anti CD19 antibodies are basically a direct cell killing through apoptosis; in fact, the binding of anti-CD20 or anti-CD19 antibodies to the CD20 or CD19 antigen can directly induce programmed cell death in B cells. Other mechanisms can be immune-mediated killing. In particular the complement-dependent cytotoxicity in which the binding of the antibody to CD20 or CD19 can activate the complement system, a part of the immune system that enhances the ability to clear pathogens and damaged cells. This activation results in the formation of the membrane attack complex, which creates pores in the B-cell membrane. The formation of these pores leads to the osmotic lysis and death of the B cell. Also remember the antibody-dependent cellular cytotoxicity (ADCC), once the anti-CD20 or anti-CD19 antibodies bind to the B cell, they can recruit immune cells, such as natural killer (NK) cells, which recognize the antibody through their FcγIII receptors. This interaction triggers the release of cytotoxic granules from NK cells leading to the lysis of the target B cell [49]. BAFF, also known as B lymphocyte stimulator, is a member of the TNF family. It is produced by various cell types, including monocytes, dendritic cells, and neutrophils, and it plays a vital role in the development and maintenance of B-cells. Anti-BAFF drugs bind to soluble BAFF, preventing it from binding to its membrane receptors BAFF-R, TACI, and BCMA, inhibiting cell proliferation and survival [42] (Fig. 1).
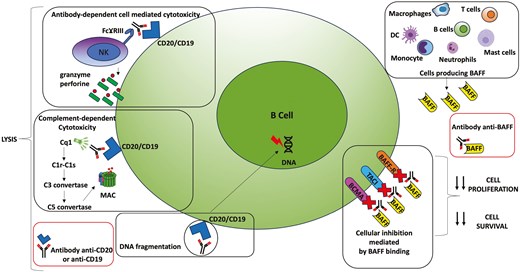
schematic figure of the mechanisms of action of anti-CD20, anti-CD19 and anti-BAFF drugs
Rituximab
Rituximab (RTX) is a mouse-human chimeric anti-CD20 monoclonal antibody which is able to deplete pre-B cells to memory B cells through ADCC [48]. In 2008, RTX was administered for the first time to 15 dcSSc patients, leading to depletion of circulating and dermal B cells [50, 51]. Since then, its effect on skin involvement was investigated by three open label trials: a study on eight patients with dcSSc treated with rituximab and methylprednisolone found effective B-cell depletion in all subjects (<5 CD19+ cells/µl blood) [48] and, at the 2 years follow-up, clinically significant improvement of modified Modified Rodnan skin score (mRSS) was evident (24.8 at baseline vs 13.6 at month 24) [52]. A following open label trial on nine SSc patients with progressive cutaneous skin involvement treated with two infusions of RTX and methylprednisolone, demonstrated a mRSS median improvement of 43.3% at 6 months together with a reduction of IL-6 levels, most likely contributing to skin fibrosis improvement [42, 53]. A French retrospective study and pooled literature analysis confirmed a potential RTX efficiency dcSSc, especially in skin and interstitial lung involvement [54].
In a 24-month, randomized, double-blind, placebo-controlled, single-centre trial, RTX was administered to eight patients (placebo = 8) with diagnosis of SSc within 2 years before enrolment [55]. Although the anti-CD20 treatment was determined to be safe for subjects with early SSc, there was no notable variance in mean skin score over time between groups. While treated patients showed some improvement in predicted FVC% and extent of lung involvement, the changes were not statistically significant. Additionally, the analysis of B cells in peripheral blood and skin tissue showed persisting B cells in skin, possibly reflecting unaffected long-lived B-cell populations, and almost complete B-cell depletion in peripheral blood but persistence of the plasmablast compartment.
More recently, the DESIRES double-blind placebo-controlled trial in 56 dcSSc or lcSSc patients confirmed the RTX efficacy on skin involvement and a favourable safety profile: the primary endpoint at 24 weeks was met in the RTX arm with a decrease in mRSS of −6.30 versus 2.14 in the placebo group [51]. Notably, as a secondary endpoint, the predicted vital capacity (FVC%) of the RTX -treated subjects, significantly improved. In a single-centre cohort study, 29 subjects previously enrolled in the DESIRES trial, continued to receive RTX once per week for 4 weeks every 6 months, showing a significantly improved mRSS and FVC% predicted at a median follow-up of 96 weeks [56]. At the last follow-up, more than 30% of patients had reduced serum IgM, IgA or IgG. Low IgM were associated with a greater improvement of predicted FVC% [57]. A beneficial role of RTX therapy in SSc-related skin and lung involvement was also suggested by a literature review by Yoshifuji et al., especially in anti-topoisomerase I-positive patients [58].
Regarding SSc related interstitial lung disease (SSc-ILD), a meta-analysis on 20 studies, including two randomized controlled trials [59, 60], concluded that RTX was associated with an improvement of both FVC and diffusion capacity at 6 months, without further increase of diffusion at 12 months [61]. Rituximab appeared to be superior in terms of FVC compared with other immunosuppressive drugs at 6 months and was associated with lower ORs of infectious adverse events. On the other hand, control immunosuppressants were different in the studies and a longer follow-up and more RCTs were recommended to avoid bias. Recently, a systematic review and meta-analysis found that, at 24–48 weeks, RTX significantly improved the FVC% predicted and the mRSS. However, it is important to note that the quality of evidence supporting these findings was very low according to the GRADE approach [62]. Latest American Thoracic Society Clinical Practice Guideline on the treatment of SSc-ILD [63], conditionally recommends RTX use of, noting that the significant reduction in disease progression and no difference in adverse events are supported by very low-quality evidence.
In patients with SSc-related pulmonary artery hypertension (PAH) rituximab was also investigated as adjuvant therapy with promising results from a double-blind, randomized, placebo-controlled trial. When data up to week 48 were included, the change in 6-minute walking test at week 24 was 25.5 ± 8.8 m for RTX compared to 0.4 ± 7.4 m for placebo, reaching statistical significance [64].
Divozilimab
Divozilimab is a humanized anti-CD20 antibody, recently approved in Russia to treat multiple sclerosis [65]. A Phase 3 randomized, double-blind, placebo-controlled clinical trial to establish the safety and efficacy of this anti-CD20 in SSc patients is currently ongoing. The primary endpoint is the change in mRSS at 24 weeks, but it will also assess the change in the FVC at week 48.
Inebilizumab
Humanised anti-CD19 monoclonal antibody inebilizumab was evaluated in a randomized, double-blind, placebo-controlled dose escalation study on 24 SSc patients [66]. Two SAEs possibly related to the drug occurred, supraventricular tachycardia and subclavian vein thrombosis, but overall, the authors conclude for a promising safety profile of the biologic agent. A potential trend of improvement in skin thickness in treated subjects but not in the small placebo group was observed, but a larger population is certainly needed to evaluate this exploratory endpoint. A Phase 3 placebo-controlled double-blind study on the superiority of inebilizumab to placebo on skin thickening is ongoing, with the first estimated primary completion date in June 2024.
Belimumab
Belimumab is an anti-BAFF human monoclonal antibody, already used in the treatment of systemic lupus erythematosus [67, 68]. BAFF, a soluble protein produced by monocytes, dendritic cells, macrophages, and other immune cells, enhances the development, survival and activity of B-cells [69]. As for SSc, belimumab was evaluated in a double-blind, placebo-controlled pilot study Gordon: 20 patients with mycophenolate mofetil as background treatment were randomized 1:1 between belimumab and placebo. Although in the belimumab group a greater median difference in mRSS was noted, it did not achieve statistical significance. Interestingly, patients with improved mRSS in the belimumab arm showed a significant decrease in expression of B cell signalling and profibrotic genes and pathways, in contrast with the placebo group [70]. In an ongoing randomized double-blind, placebo-controlled study, dcSSc patients on mycophenolate mofetil will be given a belimumab/RTX combination to evaluate the change in the ACR Composite Response Index in SSc at 12 months [71]. The deep immunosuppression derived by this treatment regimen could potentially lead to significant adverse events, an aspect that will be need a careful consideration.
Ibrutinib
Ibrutinib is an oral drug that belongs to a class of medications known as Bruton’s tyrosine kinase (BTK) irreversible inhibitors. Blocking the BTK enzyme, ibrutinib can inhibit BCR signalling by selective active site binding interfering with signals that promote the survival and proliferation of B cells, as it regulates proliferation, differentiation, and apoptosis.
In preclinical studies, ibrutinib has shown potential in modulating the immune response and reducing fibrosis, which are key features of SSc. Possible benefits of using ibrutinib in SSc could be an immune modulation that reduces the abnormal immune activity, potentially dampening the autoimmune response that drives the disease. Moreover, it could reduce fibrosis, in fact, preclinical studies suggest that ibrutinib may interfere with pro-fibrotic pathways, which could slow or reduce the tissue thickening and scarring. There is could also be anti-inflammatory effects by targeting inflammatory cells, which contributes to the disease’s progression [72]. Ibrutinib represents a promising therapeutic candidate in SSc and although preclinical studies are encouraging, direct evidence in SSc is still limited. Clinical trials will be crucial to determining its role in managing this complex disease.
Ianalumab
Ianalumab also known as ABBV-181 is an investigational monoclonal antibody that targets BAFF. This medication is primarily being studied for its potential role in autoimmune diseases, including SSc [73]. Ianalumab works by binding to BAFF, preventing it from interacting with its receptors on B cells. This inhibition leads to reduced survival and activity of autoreactive B cells. Furthermore, by targeting BAFF, ianalumab may decrease the production of autoantibodies which contribute to the inflammatory processes and tissue damage. Ianalumab represents a potential therapeutic strategy for SSc through BAFF inhibition and modulation of B cell activity. Ongoing research may clarify its role in the management of SSc and similar autoimmune conditions. A Phase 2 randomized, double-blind, parallel-group, placebo-controlled multicenter study is evaluating the efficacy, the safety and the tolerability of ianalumab in participants with diffuse cutaneous SSc.
Brentuximab vedotin
Brentuximab vedotin is an antibody-drug conjugate that combines an anti-CD30 monoclonal antibody with a cytotoxic agent, monomethyl auristatin E (MMAE). Its potential role in SSc is not well established, but some aspects of its mechanism of action may offer insights into possible therapeutic benefits. It binds specifically to the CD30 receptor, which is expressed on certain activated B cells and Reed-Sternberg cells in Hodgkin lymphoma. Upon binding, the drug is internalized, leading to the delivery of the cytotoxic agent MMAE into the target cell. MMAE disrupts the microtubule network within the cell, leading to cell cycle arrest and apoptosis [74]. Exploratory studies might investigate the broader implications of CD30 signalling in autoimmune diseases, including SSc, but this is an emerging area of research. An open-label Phase 2 study of Brentuximab vedotin for skin involvement in refractory diffuse cutaneous SSc is ongoing.
Autoantibody-targeted therapies
Intravenous immunoglobulin
Currently, intravenous immunoglobulins (IVIG) are commonly used to treat various autoimmune diseases, although their mechanism of action remains not completely understood [75]. Multiple pathways are likely involved in their immune modulatory activity and antifibrotic properties ranging from inhibition of T-cell proliferation and activation, inactivation of TNF-a and IL-1α, inhibition and modulation of phagocytosis and apoptosis, but they are also able to enhance the catabolism of autoantibodies produced by autoreactive B-cells [76–79] (Fig. 2).
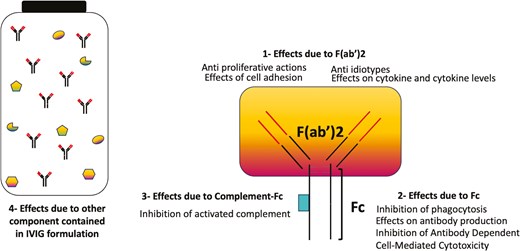
immunomodulatory actions of IVIG. (IVIg) is made up of four components responsible for immunomodulation: (i) actions mediated by the F(abʹ)2 variable regions (ii), actions of Fc on Fc receptors (FcR) (iii), actions promoted by complement binding within the Fc fragment and (iv) substances other than antibodies in IVIg preparations (cytokines, cytokines receptor, sugars, stabilizing agents) can contribute, in synergy with immunoglobulins, to immunomodulate the immune response, although the mechanisms are still to be clarified
Recently, a retrospective multicenter observational study conducted on 78 SSc patients treated with IVIG found significant improvement in muscular and skin involvement, in terms of modified manual muscle strength scale (MRC), creatine kinase (CK) levels, and mRSS. Gastrointestinal symptoms also improved. No significant changes in pulmonary function tests were observed. Patients were treated with a median of five monthly cycles of IVIG therapy (0.4 g/kg/day for 5 consecutive days) [80].
Previously, a French study on 46 SSc patients had also observed a significant improvement of musculoskeletal involvement (muscle pain and creatine kinase levels), digestive tract symptoms (gastro-oesophageal reflux disease and bowel symptoms), and systemic inflammation (CRP levels). IVIG treatment, administered at 2 g/kg/cycle was found to be well tolerated, with two serious adverse events (a deep vein thrombosis and a diffuse oedematous syndrome) [77]. The effect of IVIG treatment in SSc-associated myopathy was also addressed in a retrospective study on 52 patients. Subjects treated with IVIG (n = 18) were mainly treated in combination with corticosteroids. Myopathy remission was achieved in all subjects receiving IVIG, differently from patients who did not receive the therapy. On the other hand, no significant difference in skin or pulmonary involvement was observed [81].
Regarding skin involvement, data from real life experience on 24 patients with refractory diffuse SSc skin involvement treated with IVIG (2 g/kg body weight for 4 consecutive days monthly) showed a statistically significant mRSS improvement in 19 patients (average mRSS of −6.61 ± 5.2, P < 0.001) at 6 months and a milder improvement at 12 months (−11.45 ± 9.63, P < 0.002). On the other hand, patients with a low baseline mRSS remained stable, without significant skin improvement. The systematic review on a total of 183 patients underlined a significant benefit of IVIG treatment on skin involvement, with a suggested dose of 2 g/kg/month [82].
Recently, in three patients with an overlap syndrome SSc-myositis. Kazuki et al. described a dramatic improvement of gastrointestinal symptoms after treatment with IVIG [83]: these patients successfully left total parenteral nutrition after two cycles of IVIG and they started to gain weight. The authors suggested that there might be novel autoantibodies targeting nucleolar antigens associated with severe gastrointestinal symptoms or to a positive response to IVIG treatment, since one of the subjects had ANA positivity with nucleolar pattern without any specific autoantibody. This observation highlights the possible pivotal role of the autoantibody depleting mechanism of IVIG.
Novel approaches for the removal of antibodies
The neonatal Fc receptor (FcRn) is involved in the transport of maternal IgG to the foetus, but it also modulates the transport of antibodies and other serum proteins by binding IgG in endosomes and releasing IgG at neutral pH, enabling IgG recycling [84, 85]. Consequently, FcRn reduces lysosomal IgG degradation and regulating IgG concentrations. The inhibition of this pathway is a novel strategy to reduce autoantibodies concentrations in autoimmune diseases. Efgartigimod is the first approved neonatal Fc receptor antagonist for the treatment of generalized myasthenia gravis with acetylcholine receptor (AChR) antibody positivity. In these patients, efgartigimod was well tolerated and reduced pathogenic antibodies leading to significantly diminish disease severity [86, 87].
Several studies are being conducted to assess its efficacy and safety in the treatment of other autoimmune disease, namely pemphigus vulgaris and foliaceus [88, 89] and immune thrombocytopenic purpura [90], with promising results. At the same time, other FcRn antagonist are in Phases 1 and 2 studies, such as Rozanolixizumab and Batoclimab in myasthenia gravis [91, 92] and Orilanolimab in pemphigus [93].
Actually, a study is ongoing on this antibody depleting strategy in an SSc trial.
The new era of cellular therapies
Cellular therapies
Cellular therapies involve the administration of immune living cells to a patient to treat or cure a disease [94]. These immunotherapies are part of the broader field of regenerative medicine and have shown promise in treating a variety of cancer diseases [95]. In particular, there are six FDA-approved CAR-T cell therapies for hematological cancer. Because of the high success of CAR-T cell therapy in treating cancer, other immune cells, such as NK cells, B cells, macrophages, and dendritic cells, genetically modified could be used to treat tumours and other disease [96]. Some CAR-T cell therapies are now also being used to treat autoimmune diseases [97].
Chimeric antigen receptor-T and CAAR-T cell therapies
Chimeric antigen receptor (CAR) is a chimeric molecule which consists of an extracellular domain binding a target cell, and an intracellular signalling domain, activated upon antigen-binding [47, 97]. This receptor can direct transduced cells to a specific target (Fig. 3A). Very recently, CAR-T cells targeting CD19 have been used in autoimmune disease, obtaining a rapid B-cell depletion which persisted in time, paralleled by clinical remission (Fig. 3C). This approach has been successful in systemic lupus erythematosus [98] and antisynthetase syndrome patients [99], and very recently also in SSc. In fact, autologous CD19-directed CAR-T cells were successfully employed in a severe, refractory SSc, resulting in a fast improvement of skin and internal organs (heart, joint) [100]. The infusion was well tolerated, with just a grade 1 Cytokine Release Syndrome, and the full B-depletion was reached in a week time. Remarkably, at 3 and at 6 months anti-RNAP3 antibodies were negative. In a follow-up case series (median follow-up 15 months), four SSc patients treated with CAR-T showed a decrease of the EUSTAR activity index as well as of the severity of skin and lung involvement [101].
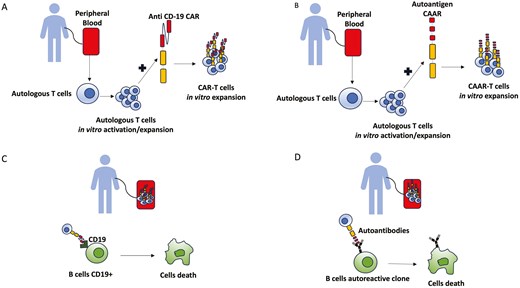
brief description of autologous CAR-T (A) and CAAR-T cells generation (B) and their mechanism of action after reinfusion (C and D)
Another interesting outlook on novel approach in autoimmune connective tissue diseases is represented by the chimeric autoantibody receptor (CAAR) therapeutic strategy (Fig. 3B). While CAR-T cells deplete both normal and autoreactive B-cells, CAAR-T are more effective and safer in autoimmune disease where the pathogenetic role of specific autoantibodies have been established. Indeed, the autoantigen is fused to a CD137-CD3ζ signalling domain so that autoantigen-CAAR-T cells deplete specific autoreactive B cells in vivo [102] (Fig. 3D). In SSc, putative targets could be autoreactive B-cell against ATA [103], PDGF-receptor, CXCL-4, angiotensin-receptor type-1 or endothelin receptor type-A, that are involved in myofibroblast activation, microvascular damage and type 1 interferon signature [104, 105]. However, at the moment in SSc it seems unlikely that targeting just a specific clone of autoreactive B-cells may have a significant effect on a complex and still not completely understood pathogenesis.
CAR-NK cell therapy
Like CAR-T, chimeric antigen receptor NK (CAR-NK) cells are a promising field of research in the treatment of cancer diseases but also autoimmune disorders. CAR-NK cells are genetically modified NK cells that have been engineered to express chimeric antigen receptors (CARs). NK cells are innate immune cells that can directly eliminate cancer or stressed cells through a delicate balance of inhibitory and activating signals. NK cells have receptors that recognize major histocompatibility complex class I (MHC-I) molecules on the surface of normal cells. When these receptors bind to MHC-I, they send inhibitory signals that block the NK cell from activating, preventing the normal cell from being killed. If the cells do not express enough MHC-I (as is often the case in cancer cells or infected or stressed cells), the inhibitory receptors are not activated. In this scenario, the activating NK cell receptors, which recognize other stress molecules on the surface of abnormal cells, take over, leading to NK cell activation without restriction by human leukocyte antigen (HLA). CAR NK cell therapies are still an emerging field and need efforts from the scientific community to establish its safety and efficacy in the treatment of oncological and autoimmune diseases. However, we report an interesting study conducted in a humanized lupus mouse model demonstrated that using PD-L1 CAR-NK-92 cells, genetically modified to eliminate PD-1-high CD4+ T cells, resulted in decreased memory B-cell proliferation with consequent decrease in Ig secretion and cell differentiation [106]. It is also important to note that CAR-NK cells can offer some advantages over the more well-known CAR-T cells. NK cells, unlike T cells, are less likely to cause GvHD, a serious complication in which immune cells attack the patient’s own tissues. CAR-NK cells also have a shorter lifespan than CAR-T cells, which may reduce the risk of long-term side effects. It is also important to note that CAR-NK cells can be created from healthy donors and stored for immediate use, potentially making them more accessible and quicker to deploy than CAR-T cells, which must be personalized for each patient [107].
Engineered B-cell therapies: dream or reality?
Macrophages and T lymphocytes have been extensively studied for their anti-tumour potential [108], while B cells, which produce antibodies and regulate the activation of NK cells and T cells, are recently gaining ground in the research field [109]. It would be interesting to understand the response of B cells by producing specific antibodies for tumour biomarkers or biomarkers involved in autoimmune diseases. There are several reasons why B lymphocytes should be considered for cellular immunotherapy as they fulfil crucial requirements. B lymphocytes are antigen-presenting cells that can easily infiltrate the tumour site or inflammatory tissues and can selectively and specifically present antigen to CD4+ T cells and CD8+ T cells even at extremely low antigen concentration [110]. These cells are also responsible for promoting the formation and expansion of T cell memory. Inspired by the success of CAR-T, scientists should aim to exploit genetically modified B cells, enhancing their natural functions to treat various diseases. Engineering B cells using gene editing techniques such as CRISPR/Cas9, viral vectors or transposons could lead to B cells producing specific, high-affinity antibodies against tumour cells, infectious agents or self-antigens [111–113]. One could imagine using the methods described above to engineer CAR-B cells capable of expressing chimeric antigen receptors that would enable them to produce specific antibodies or perform other immune functions, such as secreting immunomodulatory cytokines in a highly targeted manner. This approach would combine the antigen-specific targeting ability of CARs with the natural antibody-producing ability of B cells (Fig. 4).

schematic of steps of engineering techniques to obtain autologous CAR-B cells (A) and presumed mechanisms of action of the engineered B cells (B): 1–CAR-B cells could be engineered to produce specific antibodies against targeted antigens, enhancing the immune response against cancer cells or autoimmune markers, 2–CAR-B cells could present antigens more effectively to T cells, boosting the adaptive immune response, 3–CAR-B cells could be programmed to secrete immunomodulatory cytokines that modulate the immune environment, enhancing anti-tumour activity or reducing inflammation
In fact, edited B cells can be engineered to present specific antigens more effectively, enhancing T cell activation and immune response.
In addition, B cells could be engineered to secrete therapeutic cytokines that can modulate the immune environment, supporting immune responses or reducing inflammation. Finally, they can be generated from peripheral blood, are insensitive to immunosuppressive microenvironments, do not induce self-tolerance and B-cell immunotherapy is generally considered safer because they cause little toxicity.
Conclusions
Solid evidence shows that B-cells play a pivotal role in SSc pathogenesis, in particular in the interplay between immune system (disimmunity), vessels (vasculopathy) and extracellular matrix (fibrogenesis). However, the plasticity of the immune system, the clinical heterogeneity and the disease evolution suggest a tailored and combined treatment approach which could address the multiple SSc pathogenic mechanisms. The contribution of B cells to the disease can be approached through B-cell depleting therapies, that represent an area of significant interest. Recently, the SSc treatment paradigm has been consistently modified, and new avenues have been disclosed. The transfer of CAR-engineered T cells has developed in the field of autoimmune rheumatic diseases. In fact, further studies are warranted to confirm the role of either CAR-T and CAAR-T cell therapies as well as autoantibody targeted therapies. Further efforts are needed to increase studies on the multiple potential of engineered B cells as well as on the discovery of new biomarkers responsible for the most common pathologies such as also autoimmune diseases. Collaboration between immunologists, geneticists, and clinicians is essential to translate these innovations from the lab to clinical practice.
Abbreviations
- BAFF
B-cell activating
- Bregs
B regulatory cells
- CAR
chimeric antigen receptor factor
- IVIG
intravenous immunoglobulins
- NK
natural killer
- RTX
rituximab
- SSc
systemic sclerosis
- SSc-ILD
SSc-related interstitial lung disease
- TNF
tumour necrosis factor.
Conflict of interest
None declared.
Funding
This research received no external funding.
Author contributions
Cristina Scaletti (Conceptualization, Data curation, Supervision, Writing—original draft), Sara Pratesi (Data curation, Visualization, Writing—original draft), Silvia Bellando Randone (Writing—review & editing), Linda Di Pietro (Writing—review & editing), Corrado Campochiaro (Writing—review & editing), Francesco Annunziato (Writing—review & editing), and Marco Matucci Cerinic (Conceptualization, Supervision)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}