





Abstract
The effect of beta-adrenergic stimulation on human labial minor salivary gland epithelial cells (LMSGEC) on IL-6 production and its dependency on endoplasmic reticulum (ER) stress were investigated. Primary LMSGEC from Sjögren’s syndrome (SS) patients and controls in culture were stimulated with epinephrine and IL-6 expression was evaluated by qPCR and ELISA. The expression of β-ARs in cultured LMSGEC was tested by qPCR, while adrenoceptors and cAMP levels were examined in LMSGs by immunofluorescence. ER evaluation was performed by transmission electron microscopy (TEM) and ER stress by western blot. Epinephrine-induced IL-6 production by cultured LMSGEC was evaluated after alleviation of the ER stress by applying tauroursodeoxycholic acid (TUDCA) and silencing of PKR-like ER kinase (PERK) and activating transcription factor 4 (ATF4) RNAs. Expression of IL-6 by LMSGEC was upregulated after β-adrenergic stimulation, while the silencing of adrenoreceptors downregulated IL-6. The amelioration of ER stress, as well as the silencing of PERK/ATF4, prevented epinephrine-induced upregulation of IL-6. Adrenergic stimulation led to higher and sustained IL-6 levels secreted by LMSGEC of SS patients compared to controls. Adrenergic signaling was endogenously enhanced in LMSGEC of SS patients (expression of β-ARs in situ, intracellular cAMP in cultured LMSGEC). In parallel, SS-LMSGEC expressed dilated ER (TEM) and higher levels of GRP78/BiP. PERK/ATF4 pathway of the ER stress emerged as a considerable mediator of adrenergic stimulation for IL-6 production by the LMSGEC. An enhanced endogenous adrenergic activation and a stressed ER observed in SS-LMSGEC may contribute to a sustained IL-6 production by these cells after adrenergic stimulation.

Introduction
The immune system, in both organ-specific and systemic autoimmune diseases, shows a predilection for a target cell or a specific tissue. Different groups of investigators in the last 50 years have pointed out the significance of the target cell as the initiator and perpetuator of autoimmune diseases, in contrast to the classic theory which supports the notion of self-tolerance breakdown [1]. It is now clear that the target cells not only share common intrinsic features that renders them susceptible to autoimmune attack but also have the capacity to regulate inflammation by activating T cells and releasing cytokines and chemokines [2].
Interleukin-6 (IL-6) has emerged as one of the major mediators that govern the onset and progression of immune-mediated tissue injury [3]. In the “local initiator model” [4], injury, infection, stressors, and age-related events drive IL-6 production by non-immune cells, including epithelial cells. The released IL-6 enhances the production of other pro-inflammatory cytokines and chemokines, leading to the immune cell recruitment into the affected tissue. The cross-talk between non-immune and immune cells, particularly the activated T cells, further augments IL-6 production [4].
Sjögren’s syndrome (SS) is a systemic autoimmune disease characterized by immune-mediated destruction of salivary and lacrimal glands [5]. Studies from primary cultures have revealed that cultured labial minor salivary gland epithelial cells (LMSGEC) of SS patients retain, in vitro, an activated phenotype—as demonstrated by the inappropriate expression of immunoregulatory molecules—are able to activate T cells [6–8] and release Ro/SSA and La/SSB autoantigens through exosomes and apoptotic blebs [9, 10]. The above described metamorphosis of LMSGEC highly suggests that these cells may have a leading role in autoimmune reactivity in SS [5, 11, 12].
Despite extensive research, the precise factors, extrinsic or intrinsic, rendering the LMSGEC immunogenic have not been identified. Accumulative clinical data showed that stress and more specifically chronic, intrinsic (anxious personality), [13] or extrinsic (chronic stressful life events or posttraumatic stress) [14–16] may influence the development and exacerbation of autoimmune diseases. SS patients have higher scores of anxiety, introverted hostility, and neurotic personality compared to healthy controls and cancer patients [17], while similar personality features are also detected in patients with rheumatoid arthritis (RA) [18]. Furthermore, SS patients cannot handle effectively stressful events that usually precede the disease onset [19].
Chronic stress leads to excessive sympathetic and deficient parasympathetic tone activation [20]. This imbalance has also been described in patients with RA [21]. Epinephrine is one of the hormones whose release is enhanced during stress activating both β1- (β1-AR) and β2-adrenergic receptors (β2-AR) [22, 23]. Adrenergic stimulation has a vital role in the secretion of salivary proteins [24]. The central organelle, involved in the quality control of the continuous protein production, is the endoplasmic reticulum (ER) which is characteristically extended in secretory cells [25]. Conditions that disrupt ER homeostasis can lead to ER stress. As a consequence, an adaptive signaling pathway known as the “Unfolded Protein Response” (UPR) is activated to relieve ER stress [26].
Adrenergic stimulation of different cell types leads to ER stress, even to apoptosis, if the stimulus is intense [27]. On the other hand, in some cell types, such as fibroblasts, β-adrenergic stimulation drives IL-6 production [28]. The induction of IL-6 synthesis via adrenergic stimulation has never been associated with the intermediation of ER stress.
IL-6 is one of the cytokines that has not been studied, in depth, in the pathogenesis of SS. Circulating IL-6 is not elevated in patients with SS and inhibition of IL-6 via biological blocking of the receptor did not improve systemic involvement and symptoms [29]. However, IL-6 signaling is present in situ in LMSGEC of patients with SS [30] and this may play a crucial role in the onset and progression of local immune-mediated tissue injury.
In this study, the impact of β-adrenergic stimulation, on the IL-6 expression and the dependence of this expression to the ER stress was evaluated, using labial minor salivary gland (LMSG) tissue biopsies as well as cultured LMSGEC from SS patients (SS-LMSGEC) and disease controls (Ct-LMSGEC)
It was observed that β-adrenergic stimulation of LMSGEC induces IL-6 production via ER stress and especially through the PERK/ATF4 pathway of the UPR. Evidence is also provided that β-adrenergic activity and ER stress are endogenously enhanced in LMSGEC of SS patients leading to a higher and more sustained IL-6 production, creating locally an inflammatory milieu.
Materials and methods
Patients and LMSG biopsies
LMSG were obtained, under local anesthesia from 37 individuals undergoing diagnostic evaluation for sicca symptoms indicative of SS. Eighteen patients were diagnosed with primary SS, according to the American College of Rheumatology/EULAR 2016 criteria for SS [31]. The control group consisted of nineteen individuals with subjective sicca symptoms who did not fulfill the diagnostic criteria for SS and had a negative LMSG biopsy (Supplementary Table S1). None of the participants had evidence of lymphoma, sarcoidosis, or infection with HBV, HCV, or HIV. Individuals’ tissue specimens of LMSG were divided into parts for three separate processes: paraffin embedding for histopathologic evaluation, fixation for electron microscopy, and tissue processing for primary epithelial cell culture and establishment of LMSGEC lines.
Primary LMSGEC cultures
Long-term non-neoplastic LMSGEC lines were established from LMSG biopsies by explant outgrowth technique and maintained in culture, as previously described [6].
Immunofluorescence staining of cultured LMSGEC
LMSGEC were cultivated using 12-well chambers at a density of 5 × 104 cells/ml and maintained for two days in a growth medium. The confluent monolayer was then fixed in 4% paraformaldehyde (PFA) for 15 min and washed three times with phosphate buffer saline (PBS). Cells were permeabilized with 0.1% Triton X-100 in Tris-buffered saline (TBS) for 10 min and then blocked with 5% normal goat serum for 15 min at room temperature. After that, LMSGEC were incubated with anti-cAMP antibody overnight at 4oC and following washing with PBS, the cells were incubated with an Alexa Fluor 488 goat anti-rabbit secondary antibody for 1 h at room temperature (Supplementary Table S2). Finally, the cells were mounted with Vectashield Antifade Mounting Medium containing DAPI (Vector Labs, USA) and visualized by a Zeiss Axio Scope A1 Upright Microscope (Carl Zeiss, Germany).
Immunofluorescence staining of paraffin-embedded tissue
Paraffin-embedded salivary gland tissues were treated and stained as previously described [7]. The antibodies used in this study are listed in Supplementary Table S2. The sections were mounted with Vectashield Antifade Mounting Medium containing DAPI (Vector Labs, USA) and visualized with a confocal laser scanning microscope or a Zeiss Axio Scope.A1 upright microscope (Carl Zeiss, Germany). Quantification of immunofluorescence intensity was performed by manual counting using Image J software (NIH, USA).
Electron microscopy
Transmission electron microscopy (TEM) was performed using small tissue fragments (~1–2 mm3) from LMSG biopsies. The tissue samples were fixed in 2.5% glutaraldehyde in 0.01 M phosphate buffer saline (PBS, pH 7.4) for 24 h at room temperature and post-fixed in 1% aqueous osmium tetroxide for 1 h at 4°C as previously described [32]. The sections were observed by a FEI Morgagni 268 transmission electron microscope (Philips/FEI, USA) and images were taken with a Morada digital camera (Olympus, Germany). The ER lumen width of LMSGEC was measured by using the freehand selection tool in Image J software (NIH, USA).
Western blot
LMSGEC, after harvesting by thrypsinization, were lysed in radio-immunoprecipitation assay buffer (RIPA) containing a protease inhibitor cocktail. Cell lysates (20 μg of protein) were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4–20% Mini-PROTEAN TGX precast gels (Biorad, USA) and transferred onto a nitrocellulose membrane. Following blocking with tris-buffered saline-0,1% Tween 20 (TBST) containing 5% non-fat milk, the membrane was incubated with primary antibodies, according to the manufacturer’s instructions. After three washes with TBST, the membrane was incubated with an HRP (horseradish peroxidase)-conjugated secondary antibody for 1 h at room temperature and then was visualized with an enhanced chemiluminescence (ECL) reagent (Thermo Fisher Scientific, USA) using the Biospectrum 515 Imaging system (UVP, USA). Equal protein loading was verified using an anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody. The antibodies used in this study are listed in Supplementary Table S2.
Adrenergic stimulation
Epinephrine (Cayman Chemical, USA) was applied to LMSGEC 24 h after the plating of 2 × 105 cells/ml at 12-well cell culture plates. To determine the optimal stimulation conditions, in a titration experiment, a range of epinephrine concentrations were added into the cultures of the cells. To define the non-toxic concentration range of epinephrine, we analyzed cell death of LMSGEC using Annexin V staining followed by flow cytometry. A final concentration of epinephrine 20 μM was chosen since that concentration did not affect cell viability.
siRNAs and transfection of LMSGEC
LMSGEC were seeded in a 24-well plate or a 12-well chamber at a density of 2 × 105 cell/ml. One day following plating, small interfering RNA (siRNA) targeting ADRB1, ADRB2, PERK/EIF2AK3, ATF4 and non-targeting control siRNA (Dharmacon-Horizon Discovery, USA) at a concentration of 25 nM each were mixed with 1.2 μl (or 0.4 μl for 12-well chamber) Lipofectamine 3000 (Invitrogen—Thermo Fisher Scientific, USA), following supplier’s recommendations. Twenty-four hours after transfection, the Opti-MEM medium containing the RNA-lipid complexes was replaced by KBM and LMSGEC was subsequently treated with 20 μΜ epinephrine for 24 h. LMSGEC were then processed for immunofluorescence or qPCR analysis. Knockdown efficiency was tested with qPCR. All siRNAs used in the study are listed in Supplementary Table S3.
Real-time polymerase chain reaction (qPCR)
Total RNA was isolated from LMSGEC using Nucleozol (Macherey-Nagel, Germany), according to the manufacturer’s protocol and quantified by a Nanophotometer (Implen, USA). cDNA was synthesized from 1 μg of total RNA using a reverse transcription kit (Takara, Japan). qPCR was performed using PowerUp Sybr Green Master Mix (Applied Biosystems, USA) on StepOnePlus real-time PCR System (Applied Biosystems, USA). Expression levels of mRNA were normalized to beta-actin. Primers used in qPCR are listed in Supplementary Table S4. Relative gene expression was calculated using the 2−ΔΔCt method.
Enzyme-linked immunosorbent assay (ELISA)
Ct- and SS-LMSGEC seeded in 12-well cell culture plates, were stimulated with 20 μΜ epinephrine for various durations of time, ranging from 12 h to 72 h. Cell culture supernatants were harvested and time course analysis of IL-6 secretion in response to stimulation was performed by ELISA (cat. # 430504, Biolegend, USA), according to the manufacturer’s protocol.
Statistical analysis
For parametric analysis the t-test (paired or unpaired) was performed, whereas Mann–Whitney test was used for non-parametric data. Statistical analysis was performed using GraphPad Prism 6. The level of significance was set at 0.05.
Patient and public involvement
Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Results
Beta-adrenergic stimulation upregulates IL-6 in LMSGEC
To investigate whether β-adrenergic stimulation can modulate the synthesis of IL-6 by LMSGEC, we treated primary LMSGEC obtained from LMSG biopsies of SS patients (SS-LMSGEC) and non-SS disease controls (Ct-LMSGEC) with 20 μM epinephrine, to stimulate β-ARs. After 24 h of stimulation, IL-6 mRNA expression levels were measured using real-time polymerase chain reaction (qPCR). As shown in Fig. 1A, adrenergic stimulation upregulates IL-6 mRNA in both SS-LMSGEC (2.96 ± 0.38, P = 0.007) and Ct-LMSGEC (1.95 ± 0.22, P = 0.012).
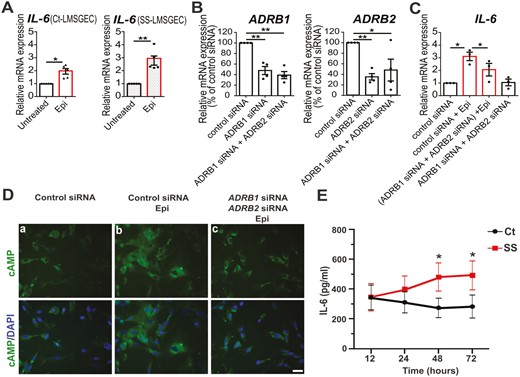
Adrenergic-mediated induction of IL-6 production by LMSGEC. (A) Relative mRNA levels of IL-6 in Ct-LMSGEC (n = 5) and SS-LMSGEC (n = 5) after stimulation with 20 μΜ epinephrine for 24 h. (B) Silencing efficiency of ADRB1 and ADRB2 in SS-LMSGEC (n = 4), based on adrenoceptors mRNA levels in LMSGEC transfected with control siRNA. (C) Relative expression of IL-6 in SS-LMSGEC (n = 3) transfected with control siRNA, ADRB1 siRNA, and ADRB2 siRNA, followed by stimulation with 20 μΜ epinephrine for 24 h. Data were normalized using ACTB mRNA levels and are represented as mean ± SEM. *P < 0.05 and **P < 0.01 by paired t-test. (D) Immunostaining of cAMP on PFA-fixed SS-LMSGEC (n = 4). Cell nuclei were visualized with DAPI. The levels of cAMP were examined in LMSGEC transfected with control siRNA, ADRB1 siRNA, and ADRB2 siRNA and were subsequently stimulated with 20 μM epinephrine for 24 h. Scale bars, 25 μm (40× magnification). (E) Time course analysis of IL-6 secretion in culture supernatants of Ct-LMSGEC (n = 6) and SS-LMSGEC (n = 5) in response to adrenergic stimulation with 20 μΜ epinephrine for 12, 24, 48, and 72 h. The results show the mean ± SEM. *P < 0.05 by unpaired t-test with Welch’s correction
To confirm the specificity of our findings and validate that β-ARs mediate the induction of IL-6 mRNA, we performed silencing of ADRB1 and ADRB2 in SS-LMSGEC using siRNAs (small interfering RNA) (25 nM each for 24 h). The efficiency of silencing was determined by qPCR, confirming a 52% reduction of ADRB1 (P = 0.006) and 65% of ADRB2 (P = 0.002) expression compared to control siRNA (Fig. 1B). Following the double silencing, SS-LMSGEC were treated with epinephrine and the expression of IL-6 was evaluated by qPCR. Our results revealed that the silencing of genes that encode β1- and β2-AR, respectively, ablates the effect of epinephrine on IL-6 induction, indicating that IL-6 upregulation in LMSGEC is mediated by β-ARs (Fig. 1C).
Since β-AR signaling activates adenylyl cyclase leading to increased cAMP [24], we monitored cAMP levels in SS-LMSGEC after silencing of ADRB1 and ADRB2. Immunofluorescence analysis confirmed that β-adrenergic signaling is attenuated under conditions of silencing (Fig. 1D).
The probability that epinephrine could induce adrenergic-mediated induction of IL-6 by increasing adrenoceptors mRNA levels, was also investigated. The results showed that mRNA levels of ADRB1, as well as ADRB2 were unaltered after stimulation of SS-LMSGEC with epinephrine, while IL-6 mRNA was upregulated (Supplementary Fig. S1).
Adrenergic stimulation leads to increased IL-6 levels secreted by SS-LMSGEC
To determine if the increased levels of IL-6 transcripts, in response to β-adrenergic stimulation, were followed by increase in IL-6 secretion, we quantified IL-6 concentration in LMSGEC culture supernatants from five patients with SS and six controls using ELISA (enzyme-linked immunosorbent assay). IL-6 levels, after adrenergic stimulation with 20 μM epinephrine, were increased at 12 h equally and rapidly in supernatants of SS- and Ct-LMSGEC (Fig. 1E). In the following time points, ranging from 24 h to 72 h, the levels of IL-6 in culture supernatants reached a plateau which was higher in SS patients compared to controls. Significant differences between Cts and SS patients were detected at 48 h (P = 0.027) and 72 h (P = 0.019) post-stimulation (2.4-fold and 2.5-fold higher levels in SS-LMSGEC, respectively) (Fig. 1E). It is noteworthy to point out that a steady-state IL-6 secretion, without epinephrine stimulation, was observed in LMSGEC which was also higher in SS-LMSGEC (Supplementary Fig. S2). The levels of IL-6 did not show any correlation with the degree of the lymphocytic infiltrates in the minor salivary gland tissue [33].
β1- and β2-adrenergic signaling is enhanced in LMSGEC of SS patients
To further examine the induction of IL-6 synthesis and secretion by LMSGEC in response to β-adrenergic stimulation, we focused on adrenergic signaling, investigating for possible differences between SS-LMSGEC and Ct-LMSGEC. To this end, we first measured the baseline mRNA expression levels of ADRB1 and ADRB2 in cultured LMSGEC from ten patients with SS and ten controls. Both transcripts, detected by qPCR, were significantly and constantly upregulated in SS-LMSGEC compared to Ct-LMSGEC (ADRB1; P = 0.007 and ADRB2; P < 0.0001) (Fig. 2A).
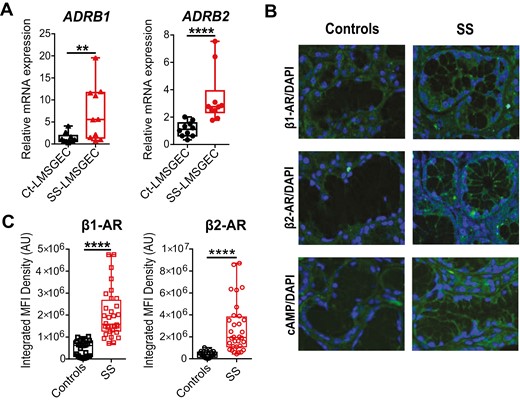
Increased β-AR signaling in LMSGEC of SS patients. (A) Baseline mRNA levels of ADRB1 and ADRB2 in cultured Ct- (n = 10) and SS-LMSGEC (n = 10). Data are represented as boxplots, with median (horizontal line), interquartile range (IQR; box), and whiskers extending to the highest and lowest values. **P < 0.01 and ****P < 0.0001 by Mann–Whitney test. (B) Representative confocal images from LMSG sections derived from Cts (n = 3) and SS patients (n = 4) stained with anti-ADRB1, anti-ADRB2 and anti-cAMP. Cell nuclei were visualized with DAPI staining (40× magnification). Control immunofluorescence without primary antibody showed no staining. (C) Quantification of immunofluorescence after β1-AR and β2-AR staining in LMSGs from Cts (n = 3) and SS patients (n = 4). Values from integrated MFI per acini are depicted as boxplots, with median (horizontal line), interquartile range (IQR; box) and whiskers extending to the highest and lowest values. ****P < 0.0001 by Mann–Whitney test
To validate this finding in situ, we analyzed the endogenous expression of β-ARs on paraffin-embedded sections obtained from LMSG biopsies of four SS patients and three Cts. The immunofluorescence staining showed that both β-AR subtypes were expressed in acinar cells of LMSGs. Both adrenoceptors were significantly higher in LMSG from SS patients compared to LMSG of Cts (Fig. 2B). Quantification of mean fluorescence intensity (MFI) further confirmed the higher density of β1-AR and β2-AR in LMSG of SS patients (P < 0.0001) (Fig. 2C). The adrenoreceptors intensity in the tissue did not show any correlation with the local intensity of the lymphocytic infiltrates nor the global grading of the infiltrates according to Tarpley’s classification [33]. The adrenoreceptors mRNA levels also did not correlate with the intensity of the lymphocytic infiltrates [33]. Taken together, our data demonstrate that independently of the inflammatory microenvironment in the LMSG lesion, the steady-state expression of β-ARs, an obvious determinant of responsiveness to catecholamines, is intrinsically enhanced in LMSGEC from SS patients.
Our findings raised the question whether the observed in situ overexpression of β1-AR and β2-AR in LMSGEC of SS patients is translated into an increased intracellular signaling in the LMSGEC of SS patients. Thus, we measured the levels of the second messenger cAMP in LMSGEC in situ, as a marker of β-AR activation. In concordance with our previous findings, immunofluorescence analysis of LMSG biopsies showed that cAMP levels were markedly increased in all glandular cells of SS patients compared to glandular cells of Cts (Fig. 2B). Our data support that the adrenergic signaling pathway is significantly enhanced in LMSGs of SS patients and this was a common finding in all tested subjects.
Dilated endoplasmic reticulum and increased expression of GRP78/BiP in LMSGEC from SS patients
Beta-adrenergic stimulation of LMSGEC drives the secretion of proteins in saliva. To achieve this, LMSGEC contains a well-developed and extensive ER, where proteins are synthesized, folded, and assembled to their native state [25]. Hence, the proper ER function and integrity are essential for the optimum-function of this cell type. To evaluate the ER status of LMSGEC in situ, we investigated its ultrastructural morphology using TEM (transmission electron microscopy). Sections of LMSG biopsies were analyzed and the ER lumen width was measured. The analysis revealed that the rough ER lumen is severely dilated in LMSGEC of SS patients in comparison with ER of Ct-LMSGEC (Fig. 3A), with a significantly higher median value of ER lumen width compared to that of Cts (P < 0.0001) (Fig. 3B). This marked dilation is a characteristic ultrastructural perturbation of ER when it is under stress [34].
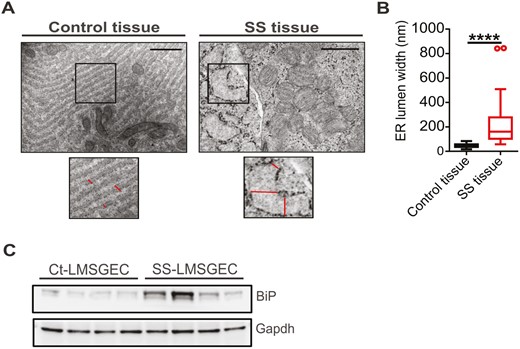
Activated ER in LMSGEC of SS patients. (A) Representative electron micrographs of SG sections from Cts and SS patients. Enlarged panels depict, at higher magnification, areas of rough endoplasmic reticulum shown in black boxes. Lines indicate the ER lumen width of LMSGEC. Scale bars; 1 μm and 500 nm for Ct- and SS-tissue, respectively. (B) Quantification of ER lumen width. Values show the median (horizontal line), interquartile range (IQR; box), and whiskers extending to the highest and lowest values, excluding outliers (>1.5*IQR; open circles). The measurements are from n = 4 subjects for each group. ****P < 0.0001 by Mann–Whitney test. (C) Western blot for analysis of GRP78/BiP expression in cell lysates of LMSGEC derived from Cts (n = 4) and SS patients (n = 4). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control
As our findings indicated that the ER of SS-LMSGEC displays a stress phenotype, it was tested whether ER stress is inherent in LMSGEC of SS patients or the inflammatory milieu acts as a stimulant to the salivary glands. Thus, we measured the baseline expression levels of major ER stress markers in cultured primary LMSGEC. Western blot analysis showed that SS-LMSGEC has increased levels of GRP78/BiP (78-kDa glucose-regulated protein/binding immunoglobulin protein) compared to those obtained from Cts (Fig. 3C), suggesting that ER perturbations may increase the demand for GRP78/BiP. Increased expression of GRP78/BiP, the ER chaperone that acts as a primary sensor for UPR activity, is a widely used marker of ER stress [35].
Amelioration of ER stress by tauroursodeoxycholic acid (TUDCA) downregulates IL-6 expression in SS-LMSGEC
TUDCA is a well-known ER stress reliever that exhibits ER chaperone-like activity, stabilizing the protein conformation and facilitating the folding capacity of ER [36]. To investigate if the ER is involved in the regulation of IL-6 expression under conditions of β-adrenergic stimulation, we evaluated IL-6 mRNA levels in epinephrine-stimulated LMSGEC in the presence and absence of 250 μΜ TUDCA for 24 h. Our results from qPCR showed that TUDCA exerts a suppressive effect on IL-6 expression and significantly prevents epinephrine-mediated induction of IL-6 in SS-LMSGEC (6.5 ± 1.2 vs. 4.0 ± 1.3, P = 0.0045) (Fig. 4A). This finding indicates that adrenergic-driven expression of IL-6 in SS patients is strongly regulated through ER.
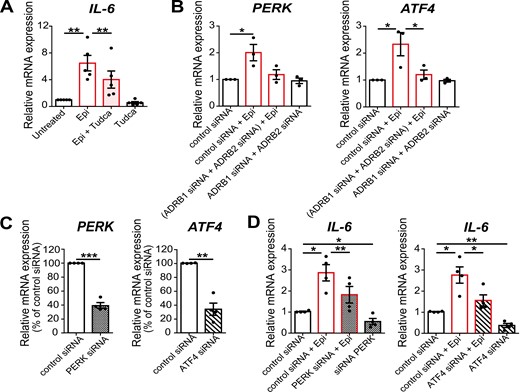
ER mediate epinephrine-induced upregulation of IL-6 in LMSGEC. (A) Relative expression of IL-6 in SS-LMSGEC (n = 5) treated with epinephrine (Epi), TUDCA (Tudca), or co-incubated with Tudca (Epi + Tudca) for 24 h (20 μM epinephrine, 250 μM Tudca). The results are expressed as fold change in treated compared to untreated SS-LMSGEC. (B) Relative expression of PERK and ATF4 in SS-LMSGEC (n = 3) after silencing of ADRB1 and ADRB2, followed by stimulation with 20 μM epinephrine. (C) Silencing efficiency of PERK and ATF4 in SS-LMSGEC (n = 4), based on mRNA levels of LMSGEC transfected with control siRNA. (D) Relative expression of IL-6 in SS-LMSGEC (n = 4) after silencing of PERK and ATF4 and subsequent stimulation with 20 μΜ epinephrine for 24 h. All data were normalized to ACTB. Symbols represent individual patient samples; bars show the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 by paired t-test.
Beta-adrenergic stimulation induces the PERK/ATF4 pathway of UPR in SS-LMSGEC
To delineate the link between adrenergic-mediated upregulation of IL-6 and ER in LMSGEC, we focused on UPR. More specifically, using the siRNA approach for ADRB1 and ADRB2 silencing in SS-LMSGEC, we assessed UPR activation status upon β-adrenergic stimulation by examining the mRNA levels of genes that encode major ER stress sensors. Among the three UPR branches, we found that epinephrine positively regulates the PERK (protein kinase R (PKR)-like endoplasmic reticulum kinase) arm, inducing the expression of PERK, as well as of downstream ATF4 (activating transcription factor 4) in SS-LMSGEC (2-fold increase, P = 0.041 and 2.3-fold increase, P = 0.045, respectively) (Fig. 4B). The double silencing efficiently blocked the epinephrine-induced upregulation of ATF4 (P = 0.042), while it was not detected statistically significant difference from PERK expression analysis (Fig. 4B). We also examined the mRNA levels of sXBP1 (spliced form of X-box binding protein 1) but no difference was detected (Supplementary Fig. S3). Collectively, our results suggest that β-adrenergic stimulation enhances the PERK/ATF4 branch of UPR.
Silencing of PERK/ATF4 pathway effectively reduces adrenergic-mediated IL-6 upregulation in SS-LMSGEC
To validate whether PERK/ATF4 pathway participates in adrenergic-mediated induction of IL-6, siRNAs targeting PERK and ATF4 were used (25 nM each for 24 h). The analysis of silencing efficiency confirmed that siRNAs effectively reduce the endogenous mRNA levels of PERK and ATF4, compared to negative control siRNA in SS-LMSGEC (60% reduction of PERK, P = 0.001 and 66% reduction of ATF4, P = 0.005) (Fig. 4C). Following the targeted silencing of PERK and ATF4, SS-LMSGEC were treated with 20 μΜ epinephrine for 24 h and subsequently the expression levels of IL-6 were determined by qPCR. As shown in Fig. 4D, silencing of PERK, as well as ATF4 resulted in a significant downregulation of epinephrine-induced IL-6 expression (PERK; 1.6-fold, P = 0.008, ATF4; 1.8-fold, P = 0.03). These findings demonstrate that IL-6 induction, upon β-adrenergic stimulation of LMSGEC, is mediated through ER activation and especially via PERK/ATF4 pathway of UPR. In parallel, we observed that in unstimulated LMSGEC, IL-6 expression was markedly decreased after silencing of both genes (PERK; P = 0.048, ATF4; P = 0.009), suggesting that PERK and ATF4 are required, at least in part, for the basal expression of IL-6 by LMSGEC (Fig. 4D).
Discussion
Four important conclusions can be drawn from this study. 1. LMSGEC are able to produce IL-6 after adrenergic stimulation. 2. An important intermediary in this process is the stress of the ER. 3. The LMSGEC of SS patients react to adrenergic stimulation with higher and prolonged production of IL-6 and 4. The ER is already in stress and functional β-adrenergic receptors are highly expressed in SS-LMSGEC than in controls.
Studies dating back to 1990 have shown that psychological and physiological stresses increase circulating levels of IL-6 in humans and animals [37, 38]. IL-6 was characterized as the “cytokine of stress” [39]. The main pathophysiological concept, for many years, was that stressors can directly affect the cells of the immune system and modulate the secretion of proinflammatory cytokines. Gradually it became clear that catecholamines may act in different, non-immune cells to induce IL-6 production [40, 41]. For example, adipose tissue releases up to 30% of circulated IL-6 [42].
Recently, Qing H et al. showed that IL-6 was the dominant cytokine induced by acute psychological stress in mice [43]. In that study, IL-6 was produced from brown adipocytes in a β3-AR-depended manner and was shown to act as a metabolic signal in hepatic gluconeogenesis. Adrenergic induction of IL-6 has also been reported by others in several cell types like astrocytes [44], hepatocytes [45], cardiac fibroblasts [46], endothelial cells [47], and macrophages [48].
In LMSGEC, adrenergic stimulation is a vital and necessary trigger of physiologic function. Catecholamines are responsible for salivary protein synthesis and secretion, through stimulation of β-ARs. However, whether catecholamines mediate IL-6 production in LΜSGEC has not previously been evaluated. Our results show that adrenergic stimulation upregulates IL-6 expression by LMSGEC. The specificity of our findings was confirmed by the double silencing of ADRB1 and ADRB2, using specific siRNAs against the two adrenoceptors.
It is well known that IL-6 is induced by stresses that are able to stress the ER [49]. LMSGEC are secretory cells and their main functional organelle is the ER. Thus, we hypothesize that ER stress and the subsequent induction of UPR can be an intermediate hub between adrenergic signaling and IL-6 production.
ER is the organelle that directs the biogenesis, folding, processing, and trafficking of membrane-bound and secreted proteins. If the protein folding capacity of the ER is disrupted or overwhelmed, the cell will initiate the UPR aiming to restore homeostasis [50]. The UPR signaling cascade is initiated by three sensor molecules; PERK, activating transcription factor 6, and inositol-requiring enzyme-1 (IRE1) to initiate downstream signaling. It lowers protein synthesis and their translocation in the ER, helping cells to adapt to the stress [51].
Multiple arms of the UPR have been reported to drive IL-6 expression. During plasma cell differentiation, IL-6 is induced through the IRE1/XBP1 arm of the UPR [52], whereas, during glucose deprivation in cancer cells, IL-6 expression is driven by a PERK/ATF4-dependent pathway [53]. PERK is a trans-ER membrane kinase that phosphorylates eukaryotic initiation factor 2α (eIF2α), which leads to selective suppression of protein translation, to enhance the correct folding of proteins and upregulation of transcription factors and chaperones such as ATF4, as a feedback protein and C/EBP homologous protein (CHOP) to initiate the apoptotic process. Meares et al. in 2014 pointed out that a PERK/JAK1/STAT3-dependent pathway drives IL-6 expression in primary astrocytes as well as other cell types in response to classic ER stress inducers [54]. In macrophages, however, the expression of ER stress-induced IL-6 is largely independent of PERK [55, 56] indicating that ER stress differently modulates IL-6 production in different cells.
In our study, it was shown, for the first time to the best of our knowledge, that adrenergic stimulation activates the PERK/ATF4 and not the IRE1/XBP1 arm of the UPR in LMSGEC. The amelioration of ER stress by TUDCA and the specific silencing of the PERK/ATF4-dependent pathway inhibited IL-6 production induced by β-adrenergic stimulation. Our data indicated that the PERK/ATF4 pathway triggered by adrenergic stimulation is involved in the induction of IL-6 from LMSGEC.
The other observation of our study was that the concentration of IL-6 secreted by LMSGEC of SS patients, after adrenergic stimulation, as it was measured in the culture supernatant, remained elevated for 72 h. This finding can be potentially linked to endogenous elevated stress of the ER of these cells. This endogenous ER stress, as indicator of cell metabolic stress, may be a fundamental component leading the SS-LMSGEC to adapt an innate immunity cell phenotype [12, 32] independently of the cause of the ER stress. We have shown that straight activation of ER stress in human submandibular gland cells causes apoptosis leading to cellular redistribution of the autoantigens Ro52 and La/SSB [57].
Following our results, we could speculate that the elevated stress of the ER can be the result of the greater presence of adrenergic receptors in SS-LMSGEC. These receptors were active and not desensitized. On the other hand, we should take into consideration that the endogenous stress of the ER in SS-LMSGEC may be responsible for energy deprivation which subsequently leads to overexpression of ARs. Overexpression of β2-ARs in mouse liver cells was linked to the metabolic status of the cells [58]. This notion is supported by the induction of IL-6 in plain culture during time without any stimulation. The only change in these culture samples is nutrient deprivation over time. The levels of glucose in the cultured cell supernatants at different time points were gradually decreased up to very low values (Supplementary Fig. S4). Another explanation can relate to the stability of IL-6 mRNA. As it has been shown in cardiac fibroblasts, β2-adrenergic stimulation, through the cAMP/PKA/CREB pathway, induces the expression of RNA-binding protein Arid5a (AT-rich interactive domain-containing protein 5A), which is a stabilizing factor for IL-6 mRNA and thus leads to elevation of IL-6 levels [40].
In conclusion, our study showed, for the first time to the best of our knowledge, that beta-adrenergic stimulation upregulates IL-6 production by human LMSGEC via ER stress. More specifically, the pathway related to PERK/ATF4, is involved in the upregulation of IL-6 in these cells. In addition, IL-6 response to adrenergic stimulation was equally acute but remained at higher levels throughout time in SS-LMSGEC compared to controls. The endogenous higher adrenergic activation and ER stress observed in SS-LMSGEC may be responsible for this phenomenon.
The results of our study raise major questions: Is the induction of IL-6 in LMSGEC by adrenergic stimulation a marker of immune alertness or a marker of cell metabolic needs? Is this phenomenon recapitulated in vivo in SG of SS patients? What is the role of the genetic predisposition? Is it worth interfering therapeutically either by blocking the beta-adrenergic axis or alleviating the ER stress in SS?
The use of human tissues, in order to observe and investigate in vitro the questions arising from clinical observations, we think that is an advantage of the study. In contrast, the human sample tissues are limited and the cultured epithelial cells are not immortalized and have limited life. This is a limitation.
Supplementary data
Supplementary data is available at Clinical and Experimental Immunology online.
Abbreviations:
- ACTB
actin beta; ADRB1: adrenoreceptor beta 1; ADRB2: adrenoreceptor beta 2; ATF4: activating transcription factor 4
- β-ARs
β-adrenergic receptors
- Ct-LMSGEC
LMSGEC from non-SS disease controls
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase; GRP78/BiP: 78-kDa glucose-regulated protein/binding immunoglobulin protein; IL-6: interleukin-6
- LMSGEC
labial minor salivary gland (LMSG) epithelial cells
- MFI
mean fluorescence intensity
- PERK
protein kinase R (PKR)-like endoplasmic reticulum kinase
- qPCR
real-time polymerase chain reaction
- SG
salivary glands; siRNA: small interfering RNA
- SS
Sjögren’s syndrome
- SS-LMSGEC
LMSGEC from SS patients; sXBP1: spliced form of X-box binding protein 1
- TEM
transmission electron microscopy
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response.
Acknowledgments
We like to thank Professor Haralampos M. Moutsopoulos for his valuable input and feedback on this project and for his continuous inspiration and guidance as well as Dr. Niki Moutsopoulos for critically reviewing the manuscript.
Ethical approval
LMSG were obtained at the Athens Euroclinic Autoimmune Diseases Department. The biopsies were obtained after informed consent and the study was approved by the hospital Bioethics Committee (approval number: 111/21032018).
Conflict of interest
The authors declare no conflict of interest.
Funding
This project has been funded by the H2020 project HARMONICSS; H2020-SC1-2016 (GA: 731944). KM was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the HFRI PhD Fellowship grant (Fellowship Number: 1642). SK was supported by a MSCA individual fellowship, H2020-MSCA-IF-2019 (GA: 897821). The funding sources had no involvement in study design; collection, analysis, and interpretation of data; decision to publish, or preparation of the manuscript.
Data availability
The data are available from the corresponding authors on reasonable request.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}