





Abstract
Transplant-associated thrombotic microangiopathy (TA-TMA) is an endotheliopathy complicating up to 30% of allogeneic hematopoietic stem cell transplants (alloHSCT). Positive feedback loops among complement, pro-inflammatory, pro-apoptotic, and coagulation cascade likely assume dominant roles at different disease stages. We hypothesized that mannose-binding lectin-associated serine protease 2 (MASP2), principal activator of the lectin complement system, is involved in the microvascular endothelial cell (MVEC) injury characteristic of TA-TMA through pathways that are susceptible to suppression by anti-MASP2 monoclonal antibody narsoplimab. Pre-treatment plasmas from 8 of 9 TA-TMA patients achieving a complete TMA response in a narsoplimab clinical trial activated caspase 8, the initial step in apoptotic injury, in human MVEC. This was reduced to control levels following narsoplimab treatment in 7 of the 8 subjects. Plasmas from 8 individuals in an observational TA-TMA study, but not 8 alloHSCT subjects without TMA, similarly activated caspase 8, which was blocked in vitro by narsoplimab. mRNA sequencing of MVEC exposed to TA-TMA or control plasmas with and without narsoplimab suggested potential mechanisms of action. The top 40 narsoplimab-affected transcripts included upregulation of SerpinB2, which blocks apoptosis by inactivating procaspase 3; CHAC1, which inhibits apoptosis in association with mitigation of oxidative stress responses; and pro-angiogenesis proteins TM4SF18, ASPM, and ESM1. Narsoplimab also suppressed transcripts encoding pro-apoptotic and pro-inflammatory proteins ZNF521, IL1R1, Fibulin-5, aggrecan, SLC14A1, and LOX1, and TMEM204, which disrupts vascular integrity. Our data suggest benefits to narsoplimab use in high-risk TA-TMA and provide a potential mechanistic basis for the clinical efficacy of narsoplimab in this disorder.
Introduction
Transplant-associated thrombotic microangiopathy (TA-TMA), defined by thrombocytopenia, microangiopathic hemolytic anemia, and organ dysfunction in the absence of disseminated intravascular coagulation, complicates 0.8–36% of allogeneic hematopoietic stem cell transplants (alloHSCT) in adults and children [1–3]. Three-year survival rates are as low as 11% [3]. Involvement of complement in the vascular injury characteristic of TA-TMA has been clearly established. In pediatric alloHSCT cohorts: activation of complement transcripts correlates with TMA development [4]; increases in circulating terminal complement complex sC5b-9 (also known as membrane attack complex (MAC)) preceded TMA development in 10/10 children vs. a rise in sC5b-9 for only 9 of 23 without TMA [5]; and 65% of the TMA patients had variants in ≥1 of 17 genes linked to promotion of complement activation, mutations that are also characteristic of an atypical hemolytic uremic syndrome (aHUS)-type of TMA [6], vs. 9% of HSCT subjects not developing a TMA [7]. Among adults post-alloHSCT, a TA-TMA diagnosis is similarly accompanied by elevated sC5b-9 levels vs. those of healthy donors, as shown by our group [8] and others [9, 10], as well as higher sC5b-9 levels among those alloHSCT patients developing a TA-TMA vs. those absent this complication post-transplant [11]. Involvement of known complement and complement regulatory genes in adult TA-TMAs is much less clear than in pediatric cohorts, however [12].
Persistent activation of both the alternative and lectin pathways (LP) of complement appears to be involved in TA-TMA. The latter may stem from binding of pattern recognition molecules such as mannose-binding lectin (MBL) to damage-associated molecular patterns (DAMPs) present on injured endothelium, activating MBL associated serine protease 2 (MASP2) to promote activation of C4, C3, and C5, ultimately resulting in the formation of C5a and C5b-9 [8, 13]. We found MASP2 levels in adult alloHSCT patients elevated over normal controls regardless of TMA development [8]. The absence of a rise in circulating MASP2 at the time of TA-TMA recognition, accompanied by a significant variance in MASP2 levels, was postulated to reflect the consumption of MASP2 on systemic microvasculature. This is supported by our recent demonstration of MASP2 and/or C5b-9 deposition on microvessels in skin or bone marrow in TA-TMA patients but not in alloHSCT patients without a TMA [12].
How these associations may translate into effective therapies for TA-TMA is an evolving issue. Anti-C5 monoclonal antibody (mAb) eculizumab has improved TMA response rates and survival in allogeneic- and autologous-HSCT TMAs in children [14, 15], but those were single institution studies and its impact in adults is much less clear, based primarily on case reports and observational studies [3, 16]. Narsoplimab, a fully humanized mAb that binds to and inhibits MASP2, was recently tested in National Clinical Trials Network Study NCT02222545, a single-arm open-label trial of 28 adult TA-TMA cases [17]. The rate of response, requiring improvement in both laboratory manifestations (platelet count and serum lactate dehydrogenase (LDH)) and organ function and/or freedom from platelet or red cell transfusions, was 61% for the full analysis set of individuals receiving at least one dose of narsoplimab [17]. One-hundred-day survival post-TMA recognition was 68% and 94% in the full analysis set and among responders, respectively [17]. A contemporary control group was not available; however, an expert review of patient severity suggests these 100-day survival data exceed that of historical cases [1, 2, 18].
We sought to determine the impact of narsoplimab in models of patient plasma-induced microvascular endothelial cell (MVEC) activation and injury developed by our group to investigate aHUS and related forms of TMA [19]. We examined plasmas from NCT02222545, the TA-TMA narsoplimab trial, and from NCT02604420, an observational adult TA-TMA [12] cohort from Weill Cornell Medicine (WCM). We employed genome-wide RNA sequencing (RNAseq) to generate hypotheses as to the putative mechanisms of action of narsoplimab in TA-TMA.
Materials and methods
Study subjects and ethical approval
Observational alloHSCT TA-TMA cohort
These individuals represent a subset of 100 adults from a prospective observational study of TMA development post-alloHSCT. It was approved by the WCM Institutional Review Board, registered with the National Clinical Trials Network (NCT02604420), and informed consent was obtained from all subjects. Characterizations of these individuals in terms of circulating MASP2 and sC5b-9 levels and MASP2 and C5b-9 deposition in the microvasculature of normal-appearing skin and bone marrow have been published [8, 12]. Inclusion criteria for a TA-TMA diagnosis consisted of five peripheral markers: microangiopathic changes, based upon a persistent increase over baseline of peripheral schistocytes; LDH levels exceeding the upper limit of normal; haptoglobin levels <50 mg/dl; de novo thrombocytopenia, based on a platelet count <50 × 109/L, or a ≥25% decrease from baseline; absence of a coagulopathy as defined by aPTT and INR within the normal range; and a negative direct Coombs test. This is consistent with a recent multi-national consensus statement regarding diagnostic criteria for TA-TMA [2]. Per protocol, it was recommended that calcineurin inhibitors (CNI) such as cyclosporine and tacrolimus and mammalian target of rapamycin inhibitors (mTORi) such as sirolimus and everolimus be held upon initial TMA diagnosis, and glucocorticoids and mycophenolate mofetil substituted. This was based on a limited literature suggesting that some TA-TMAs may respond to discontinuation of these drugs [2, 20].
Nine (9%) individuals with TMAs in the observational alloHSCT study met current harmonization criteria for a “high-risk” TA-TMA [2], having elevated levels of sC5b-9 as well as an acute kidney injury (AKI) that persisted following withdrawal of a CNI or mTORi. In addition, 2 of the 9 had grade III graft vs. host disease (GVHD). Samples were available from eight of these individuals. They were diagnosed with a median of 120 days (range 34–190 days) post-transplant. Eight additional alloHSCT patients who did not develop a TMA over a 1.5-year observation period were used as controls. They contributed plasmas collected at approximately the same times post-transplant as the TA-TMA cohort for this study.
Nasoplimab-treated TA-TMA cohort
This was a single-arm open-label trial of narsoplimab for the treatment of TA-TMA post-alloHSCT in adults (NCT02222545). Enrollment criteria, TMA definition, and response criteria were previously described [17]. Narsoplimab was administered intravenously at a dose of 4 mg/kg once weekly, based on evidence that this dosage provided >80% inhibition of MASP2 throughout the dosing interval and was well-tolerated [17]. Plasmas collected at the time of TA-TMA recognition, and for varying periods after initiation of narsoplimab administration (3–16 longitudinal samples/patient), were available to us from 13 of the 28 participants. Nine of these were TMA clinical responders and 4 non-responders. Of these 13 subjects, 12 satisfied harmonization criteria for a high-risk TMA: 8 of the 9 clinical responders and 4 of the 4 non-responders. Otherwise, no clinical or clinical laboratory-based distinctions were apparent between narsoplimab responders and non-responders. sC5b-9 levels were not available from these subjects.
Endothelial cell cultures
Primary human neonatal MVEC of dermal origin were purchased from Lifeline Cell Technology (Frederick, MD). EC was maintained in T-25 flasks (Greiner Bio-One GmBH, Germany) coated with 0.1% gelatin (Lifeline Cell Technology) using LifeFactors Vasculife VEGF-Mv medium (Lifeline Cell Technology) plus penicillin, streptomycin, and 15% fetal bovine serum. Subcultures involved a 2–5 min. Exposure to 0.25% trypsin–EDTA, followed by washing with PBS, pH 7.2. EC was used in passages 2–5.
Caspase 8 assay
MVECs, 1.5 × 105/well, were cultured in 12 well 0.1% gelatin-coated plates and incubated overnight at 37°C in serum-free medium. The next day the cultures were overlain with control or patient plasmas (2% v/v) in the presence or absence of varying concentrations of narsoplimab (also known as OMS721) or medium for ~5 h at 37°C. Caspase 8 activity was assessed using a functional assay based on the hydrolysis of acetyl-Ile-Glu-Thr-Asp-p-nitraniline, performed according to the manufacturer’s instructions (Caspase 8 Assay Kit, colorimetric; Abcam), with measurement of optical density (OD) absorbance at 405 nm.
RNA sequencing
One milliliter of 1 × 106/ml MVEC in serum-free medium was added to each well of a 6 well polystyrene culture plate coated with 0.1% gelatin and incubated overnight at 37°C. The following morning the wells were washed with PBS to remove non-adherent cells and appropriate materials were added. Each experiment involved four conditions: control plasma (i.e. plasmas from alloHSCT recipients who did not develop a TMA); TA-TMA plasma (i.e. plasmas from alloHSCT recipients who developed a high-risk TMA); narsoplimab (10 µg/ml) + control plasma; narsoplimab + TA-TMA plasma. Each condition involved a pool of plasmas from separate subjects (alloHSCT control or TA-TMA patients, three subjects each in two experiments, and two subjects each in one experiment) at a final volume of 2%. MVEC were incubated at 37°C for ~5 h and then total RNA isolated using a RNeasy Mini Kit (Qiagen). The concentration and integrity of all RNAs were determined by the WCM Genomics Core Facility using an Agilent Bioanalyzer. Poly-A libraries were prepared cDNAs generated, and gene sequencing performed in the Core facility.
Single-cell RNAseq data for cutaneous cell types were downloaded from Tabula Sapiens into R package Seurat [21]. Inference for putative cell types in our normalized bulk RNAseq samples was performed via label transfer using Seurant function TransferData with prediction scores generated per cell type in each sample.
Classification of genes upregulated or downregulated was performed by analyzing RPKM (Reads Per Kilobase of transcript per Million mapped reads) using deltaRpkm software [22] with reference to the Tabula Sapiens Reference Atlas, a vascular endothelial expression list (https://tabula-sapiens-cellxgene.ds.czbiohub.org/skin). The next generation sequencing data were then mapped to the human reference genome GRCh38 using HISAT2 (v2.2.1) aligner [23]. Gene-wise expression counts were quantified using featureCounts (v2.0.3) [24]. After filtering and quality control, Bioconductor R package edgeR (v3.38.4) [25] was used to calculate library normalized RPKM, Log2 counts/million matrices, and gene-level differential expression analysis.
Transcriptomic data analyses were summarized in the form of heat maps and gene set enrichment plots. Heat maps of significantly regulated pathways were generated using CRAN R gplots (v3.1.3) and the pheatmap function. Data exploration and dimensionality reduction involved multidimensional scaling and principal component analysis using Log2 counts/million values. An unbiased gene set enrichment analysis (GSEA) was performed using WebGestalt 2019 [26] for KEGG, Gene Ontology (GO), and WikiPathway.
Statistical analysis
Differences in plasma-based biomarkers were calculated using a two-tailed t-test in GraphPad Prism software (v9.1.0) (GraphPad, San Diego, CA). For the RNAseq experiments, the base mean, log2 change, and SE were calculated using edgeR on the pre-processed RNA-seq dataset. The enrichment score and normalized enrichment score (NES) were calculated with WebGestalt GSEA on differentially expressed genes with a significance threshold determined by deltaRKPM [22].
Data accessibility
The RNAseq data are available by contacting Jeffrey Laurence: [email protected].
Results
Longitudinal assessment of effects of plasmas from acute TA-TMA patients pre- and post-narsoplimab treatment on caspase 8 induction in MVEC
The caspase cascade is highly relevant to the pathophysiology of TA-TMA. Histologic analyses of multiple organs at autopsy document extensive apoptotic bodies in TA-TMA patients [27–30], similar to that seen in thrombotic thrombocytopenic purpura (TTP) and aHUS types of TMA [6, 31], and there is a direct correlation between TTP patient plasma-mediated caspase activation and subsequent MVEC apoptosis in vitro, as characterized by DAPI staining and DNA histograms of propidium iodide-stained cells [19]. We utilized plasmas from TA-TMA patients participating in the narsoplimab clinical trial, 9 TMA clinical responders and 4 non-responders [17], in a caspase 8 MVEC activation assay. Caspase 8 exists as a proenzyme that is converted into an active form upon recruitment to the cytoplasmic domain of activated death receptors, leading to caspase 3 induction and apoptotic injury [32]. Controls included plasmas from three adult alloHSCT patients who did not develop a TMA, obtained at about day 120 post-transplant, consistent with the median time for TMA development. These subjects were derived from the WCM observational cohort as a similar population was not part of the narsoplimab trial.
Representative longitudinal plots for 7 of the clinical responders and 3 non-responders, along with the 3 alloHSCT/no TMA controls, are illustrated in Fig. 1A and B.
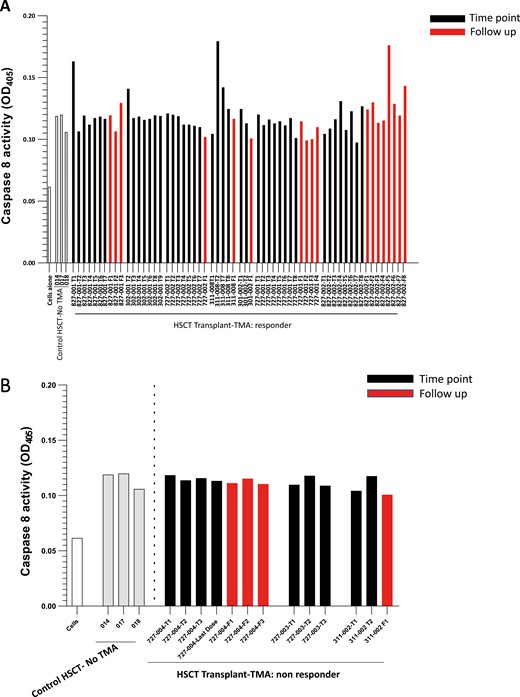
Caspase 8 activation I MVEC by plasmas from acute TA-TMA patients pre-, during, and post-narsoplimab administration. Cultures of primary human neonatal dermal microvascular endothelial cells (MVEC) were exposed to plasma (2% v/v) from patients with acute TA-TMA collected prior to (black bars, T1), during (subsequent black bars), and following (red bars) cessation of narsoplimab administration. These subjects were part of clinical trial NCT02222545. Clinical response was defined as described in the text. The grey bars represent plasmas from three patients who underwent an allogeneic stem cell transplant but did not develop a TMA. They were collected as part of an observational cohort (NCT02604420) at about day 120 post-transplant, the median time for TA-TMA development. A. Serial samples from 7 of the 9 clinical responders to narsoplimab are illustrated. B. Serial samples from 3 of the 4 clinical non-responders to narsoplimab are illustrated.
Pre-treatment plasmas from 8 of 9 TA-TMA responders induced caspase 8 activity over that seen in the alloHSCT controls. This activity was suppressed to control levels during narsoplimab treatment (Fig. 1A, black bars) and through the up-to-one-month follow-up period after drug discontinuation (Fig. 1A, red bars) in all but one of the clinical responders. (Subject 827-002 is the exception.) In a few patients, this decline in caspase 8 occurred very rapidly, after one or two doses of drug (Fig. 1A). These changes could not have been an ex vivo artifact related to narsoplimab, given that the plasma samples were collected just prior to each narsoplimab dose and further diluted in the assay, resulting in only nominal concentrations of drug in these cultures. In contrast, plasmas from none of the narsoplimab clinical non-responders activated caspase 8 (Fig. 1B).
Establishing clinically relevant concentrations of narsoplimab for use in an in vitro model of MVEC activation
We previously reported that plateau inhibition (mean 65.7%, range 36.8–99.4%) of caspase 8 activation induced by plasmas from patients TTP and aHUS types of TMA that were not linked to alloHSCT occurred at 1.2 μg/ml of narsoplimab [8]. This is consistent with pharmacokinetics in healthy human volunteers administered a single 4 mg/kg dose of narsoplimab, indicating an EC50 of 0.5 µg/ml (3.3 nM), similar to the IC50 of LP inhibition by this drug in vitro [33]. But tissue-bound drug concentrations are uncertain and likely to be much higher. We now sought to determine whether caspase 8 activation in MVEC by acute TA-TMA patient plasmas derived from the WCM observational cohort could similarly be suppressed by narsoplimab in vitro. MVEC were exposed to plasmas from a pool of three acute TA-TMA patients in the presence of 0–50 µg/ml narsoplimab and caspase 8 activation was assessed. As shown in a representative experiment, 5 µg/ml showed clear but incomplete inhibition whereas concentrations ≥25 µg/ml blocked caspase 8 activation (Fig. 2). Therefore, 10 µg/ml was utilized in all subsequent experiments.
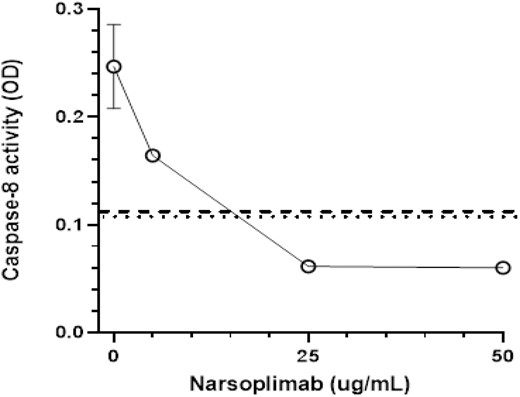
Dose–response curve for inhibition of acute TA-TMA patient plasma-mediated caspase 8 activation in MVEC in vitro. Cultures of primary human neonatal dermal microvascular endothelial cells (MVEC) were exposed to pooled plasma (2% v/v) from three patients with acute TA-TMA in the presence of varying concentrations of narsoplimab. Caspase 8 activity was analyzed in cell lysates after 5 h of incubation. Dotted and dashed lines represent caspase activity in cultures exposed to pooled plasma from healthy individuals or allogeneic hematopoietic stem cell patients who did not develop a TMA, respectively.
RNAseq reveals potential mechanisms for narsoplimab regulation of TA-TMA plasma-induced MVEC activation and injury
We utilized single-cell data from the Tabula Sapiens single-cell RNAseq skin atlas as a quality control check of the identity of our target cells as dermal MVEC. Cutaneous MVEC was the top match for normalized bulk RNAseq transcripts from each of the three replicate RNAseq experiments as described in Materials and Methods, with a predicted cell type score between 0.51 and 1.0. In subsequent analyses we pooled RNAseq results from identical conditions of the three experiments, comprising a total of 8 acute TA-TMA subjects and 8 alloHSCT patients not developing a TMA.
Distinct gene set enrichment analyses were performed referencing KEGG, GO, and WikiPathways databases. In comparing MVEC transcripts derived from cultures including TA-TMA plasmas with vs. without narsoplimab, all three pathway analyses revealed a predominance of transcripts encoding genes involved in control of the cell cycle and DNA replication and repair (Fig. 3A). Transcripts derived from cultures comparing the impact of narsoplimab in the presence of alloHSCT patient control (i.e. no TMA) plasmas also documented prominent changes related to cytokine generation and signaling linked to TNF-α, IL-1, IL-18, and interferon type II (IFN-ɣ), and to NFƙB-mediated survival signaling (Fig. 3B). Circulating levels of these cytokines correlate directly with development of the major TMAs, including aHUS, TTP, and TA-TMA [4, 19, 34].
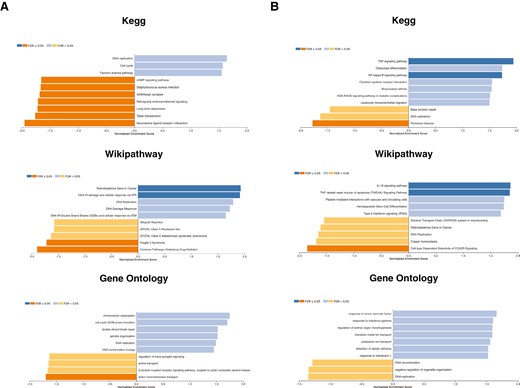
Gene set enrichment pathway analyses of MVEC transcripts from MVEC exposed to alloHSCT patient plasmas in the presence an absence of narsoplimab. A. MVEC cultures were exposed to pooled acute TA-TMA patient plasmas in the presence and absence of narsoplimab (10 µg/ml) and caspase 8 activity assessed as described in Fig. 1. All three databases highlighted effects related to genes involved in control of the cell cycle and DNA replication and repair. B. MVEC cultures were exposed to pooled plasmas from alloHSCT patients who did not develop a TA-TMA in the presence and absence of narsoplimab (10 µg/ml) and caspase 8 activity assessed. Prominent changes in cytokine generation and signaling related to TNF-α, IL-1, IL-18, and interferon type II (IFN-γ), as well as NFκB survival signaling were identified.
The importance of inflammatory pathway signaling to the effect of narsoplimab on MVEC exposed to TA-TMA plasmas was further emphasized by KEGG pathway-generated heat maps. The three most significantly affected pathways, based on RPKM scores, were cell cycle, TNF signaling, and NFκB signaling (Fig. 4A).
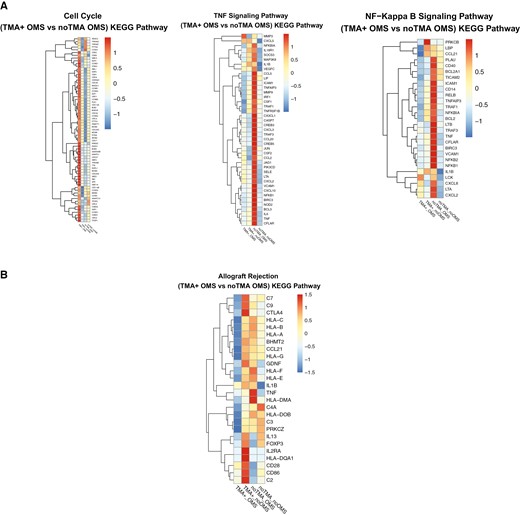
Heat maps generated by KEGG pathway and WikiPathway analysis identify specific transcripts related to narsoplimab control of MVEC activation and injury induced by TA-TMA patient plasmas. A. MVEC cultures were exposed to pooled acute TA-TMA patient plasmas in the presence and absence of narsoplimab (10 µg/ml) and RNAseq analysis performed as described in the text. KEGG pathway analysis highlighted genes involved in control of cell cycle, TNF signaling, and NFκB signaling. (“OMS” refers to narsoplimab, originally known as OMS721.) B. Further analysis of RNAs isolated from these cultures using WikiPathway illustrate changes in complement and MHC-I and -II transcripts.
In terms of complement components, WikiPathway analysis of genes involved in renal allograft rejection—a complement-activating condition that is also a trigger for an aHUS-type of TMA [6, 9]—revealed marked inhibition of transcripts for complement proteins C2, C3, C7, and C9, along with down-regulation of MHC-I and -II in narsoplimab-treated cultures (Fig. 4B).
Finally, the 20 transcripts with the greatest changes induced by narsoplimab in TA-TMA plasma-exposed MVEC in terms of upregulation, and the 20 with the greatest magnitude of downregulation, as determined by mean RPKM score irrespective of pathway association, were recorded (Table 1). Specifically:
Top 20 significant transcripts upregulated and top 20 down-regulated by narsoplimab in TA-TMA patient plasma-exposed MVEC
| Upregulated | LogFC | F | Downregulated | LogFC | F |
|---|---|---|---|---|---|
| RNU6-678P | 5.12 | 2.74 | SLC14A1 | −2.00 | 0.80 |
| MIR4766 | 4.91 | 3.75 | C2orf66 | −1.26 | 1.08 |
| MIR589 | 4.16 | 5.05 | ZNF521 | −0.42 | 2.51 |
| SNORD101 | 1.99 | 1.15 | WFS1 | −0.35 | 2.01 |
| MIR6813 | 1.98 | 0.68 | SRD5A3-AS1 | −0.33 | 0.14 |
| SERPINB2 | 1.90 | 7.81 | KRT7 | −0.30 | 0.86 |
| MIR4477B | 1.70 | 3.38 | IL1R1 | −0.28 | 1.44 |
| CHAC1 | 1.64 | 9.59 | FBLN5 | −0.27 | 3.73 |
| MT1L | 1.60 | 3.94 | SOX18 | −0.25 | 1.67 |
| TM4SF18 | 1.53 | 34.21 | ACAN | −0.21 | 0.36 |
| ASPM | 1.43 | 22.57 | RAB11FIP1 | −0.21 | 1.21 |
| CA2 | 1.42 | 7.41 | JUP | −0.20 | 2.51 |
| MIR762 | 1.41 | 2.03 | PPP1R10 | −0.20 | 1.15 |
| PSAT1 | 1.35 | 25.88 | ALPK3 | −0.20 | 2.48 |
| SPC25 | 1.33 | 8.45 | HOXD9 | −0.20 | 2.00 |
| SNCB | 1.33 | 11.87 | MIR5587 | −0.20 | 0.04 |
| TNFSF15 | 1.32 | 12.53 | TMEM204 | −0.19 | 1.60 |
| ESM1 | 1.26 | 13.92 | DOCK9 | −0.18 | 1.16 |
| TTK | 1.26 | 8.77 | BICD1 | −0.18 | 1.53 |
| TNFSF18 | 1.23 | 9.34 | LOX | −0.18 | 0.70 |
| Upregulated | LogFC | F | Downregulated | LogFC | F |
|---|---|---|---|---|---|
| RNU6-678P | 5.12 | 2.74 | SLC14A1 | −2.00 | 0.80 |
| MIR4766 | 4.91 | 3.75 | C2orf66 | −1.26 | 1.08 |
| MIR589 | 4.16 | 5.05 | ZNF521 | −0.42 | 2.51 |
| SNORD101 | 1.99 | 1.15 | WFS1 | −0.35 | 2.01 |
| MIR6813 | 1.98 | 0.68 | SRD5A3-AS1 | −0.33 | 0.14 |
| SERPINB2 | 1.90 | 7.81 | KRT7 | −0.30 | 0.86 |
| MIR4477B | 1.70 | 3.38 | IL1R1 | −0.28 | 1.44 |
| CHAC1 | 1.64 | 9.59 | FBLN5 | −0.27 | 3.73 |
| MT1L | 1.60 | 3.94 | SOX18 | −0.25 | 1.67 |
| TM4SF18 | 1.53 | 34.21 | ACAN | −0.21 | 0.36 |
| ASPM | 1.43 | 22.57 | RAB11FIP1 | −0.21 | 1.21 |
| CA2 | 1.42 | 7.41 | JUP | −0.20 | 2.51 |
| MIR762 | 1.41 | 2.03 | PPP1R10 | −0.20 | 1.15 |
| PSAT1 | 1.35 | 25.88 | ALPK3 | −0.20 | 2.48 |
| SPC25 | 1.33 | 8.45 | HOXD9 | −0.20 | 2.00 |
| SNCB | 1.33 | 11.87 | MIR5587 | −0.20 | 0.04 |
| TNFSF15 | 1.32 | 12.53 | TMEM204 | −0.19 | 1.60 |
| ESM1 | 1.26 | 13.92 | DOCK9 | −0.18 | 1.16 |
| TTK | 1.26 | 8.77 | BICD1 | −0.18 | 1.53 |
| TNFSF18 | 1.23 | 9.34 | LOX | −0.18 | 0.70 |
Top 20 significant transcripts upregulated and top 20 down-regulated by narsoplimab in TA-TMA patient plasma-exposed MVEC
| Upregulated | LogFC | F | Downregulated | LogFC | F |
|---|---|---|---|---|---|
| RNU6-678P | 5.12 | 2.74 | SLC14A1 | −2.00 | 0.80 |
| MIR4766 | 4.91 | 3.75 | C2orf66 | −1.26 | 1.08 |
| MIR589 | 4.16 | 5.05 | ZNF521 | −0.42 | 2.51 |
| SNORD101 | 1.99 | 1.15 | WFS1 | −0.35 | 2.01 |
| MIR6813 | 1.98 | 0.68 | SRD5A3-AS1 | −0.33 | 0.14 |
| SERPINB2 | 1.90 | 7.81 | KRT7 | −0.30 | 0.86 |
| MIR4477B | 1.70 | 3.38 | IL1R1 | −0.28 | 1.44 |
| CHAC1 | 1.64 | 9.59 | FBLN5 | −0.27 | 3.73 |
| MT1L | 1.60 | 3.94 | SOX18 | −0.25 | 1.67 |
| TM4SF18 | 1.53 | 34.21 | ACAN | −0.21 | 0.36 |
| ASPM | 1.43 | 22.57 | RAB11FIP1 | −0.21 | 1.21 |
| CA2 | 1.42 | 7.41 | JUP | −0.20 | 2.51 |
| MIR762 | 1.41 | 2.03 | PPP1R10 | −0.20 | 1.15 |
| PSAT1 | 1.35 | 25.88 | ALPK3 | −0.20 | 2.48 |
| SPC25 | 1.33 | 8.45 | HOXD9 | −0.20 | 2.00 |
| SNCB | 1.33 | 11.87 | MIR5587 | −0.20 | 0.04 |
| TNFSF15 | 1.32 | 12.53 | TMEM204 | −0.19 | 1.60 |
| ESM1 | 1.26 | 13.92 | DOCK9 | −0.18 | 1.16 |
| TTK | 1.26 | 8.77 | BICD1 | −0.18 | 1.53 |
| TNFSF18 | 1.23 | 9.34 | LOX | −0.18 | 0.70 |
| Upregulated | LogFC | F | Downregulated | LogFC | F |
|---|---|---|---|---|---|
| RNU6-678P | 5.12 | 2.74 | SLC14A1 | −2.00 | 0.80 |
| MIR4766 | 4.91 | 3.75 | C2orf66 | −1.26 | 1.08 |
| MIR589 | 4.16 | 5.05 | ZNF521 | −0.42 | 2.51 |
| SNORD101 | 1.99 | 1.15 | WFS1 | −0.35 | 2.01 |
| MIR6813 | 1.98 | 0.68 | SRD5A3-AS1 | −0.33 | 0.14 |
| SERPINB2 | 1.90 | 7.81 | KRT7 | −0.30 | 0.86 |
| MIR4477B | 1.70 | 3.38 | IL1R1 | −0.28 | 1.44 |
| CHAC1 | 1.64 | 9.59 | FBLN5 | −0.27 | 3.73 |
| MT1L | 1.60 | 3.94 | SOX18 | −0.25 | 1.67 |
| TM4SF18 | 1.53 | 34.21 | ACAN | −0.21 | 0.36 |
| ASPM | 1.43 | 22.57 | RAB11FIP1 | −0.21 | 1.21 |
| CA2 | 1.42 | 7.41 | JUP | −0.20 | 2.51 |
| MIR762 | 1.41 | 2.03 | PPP1R10 | −0.20 | 1.15 |
| PSAT1 | 1.35 | 25.88 | ALPK3 | −0.20 | 2.48 |
| SPC25 | 1.33 | 8.45 | HOXD9 | −0.20 | 2.00 |
| SNCB | 1.33 | 11.87 | MIR5587 | −0.20 | 0.04 |
| TNFSF15 | 1.32 | 12.53 | TMEM204 | −0.19 | 1.60 |
| ESM1 | 1.26 | 13.92 | DOCK9 | −0.18 | 1.16 |
| TTK | 1.26 | 8.77 | BICD1 | −0.18 | 1.53 |
| TNFSF18 | 1.23 | 9.34 | LOX | −0.18 | 0.70 |
MicroRNA (miRNA) regulation
Virtually all miRNAs, transcripts involved in post-transcriptional regulation of gene expression by influencing the stability and translation of mRNAs, that were modulated by narsoplimab would result in the capacity to suppress pro-inflammatory cascades and apoptosis. Most of these processes involved miRNA upregulation. For example, miR6813 is an anti-apoptotic transcript that is widely expressed in tissues. Its inhibition leads to induction of apoptosis in malignant cells [35]. miR-4766 suppresses chemokine CXCL5 [36]. CXCL5 is involved in promotion of CXCR2-dependent neutrophil trafficking and neutrophil extracellular trap (NET) formation, facilitating an acute inflammatory response, and in ischemia-reperfusion injuries linked to complement activation [37, 38]. miR-589 reverses oxidative stress and dysfunction in MVEC by targeting early growth response-1 (EGR-1) [39]. EGR-1 is a central transcription factor in EC biology, controlling expression of >300 genes, including pro-inflammatory cytokines and tissue factor, a key part of the coagulation cascade [40].
Consistent with those changes related to augmentation of miRNA transcripts, miR5582 was downregulated in narsoplimab-treated cultures. Upregulation of this miRNA promotes apoptosis and cell cycle arrest through direct targeting of CDK2 and signal adapter proteins GAB1 (growth factor receptor bound 2 associated binding 1) and SHC1 (src homology 2 domain-containing 1) [41].
Upregulation of miR762 was the only exception to the hypothesized direction of miRNA transcript modulation in protection of MVEC against pro-apoptotic, pro-inflammatory insults. Its upregulation is associated with an increase in reactive oxygen species (ROS) levels and apoptotic cell death through the regulation of ND2, a core assembly subunit of mitochondrial complex 1 [42].
Narsoplimab-associated upregulation of transcripts for functional proteins
The serine protease inhibitor SerpinB2 (also known as plasminogen activator inhibitor-2 (PAI-2)) is a dominant stress response protein. It is induced to help cells accommodate myriad stress signals, including those related to cytokine signaling, ROS production, and inflammation [43, 44]. It protects cells against TNF-mediated apoptosis by interacting with transglutaminase 2 (TG2) to inactivate procaspase 3 [45].
Procaspase 3 is also blocked by the dual-specificity type kinase (TTK, also known as mucopolysaccharidase 1 (Msp1)) acting through phosphorylation of c-Abl, preventing c-Abl from nuclear transport to limit apoptotic cell death induced by a variety of oxidative stressors [46, 47]. Narsoplimab upregulated TTK in concert with SerpinB2. Similarly, CHAC1 (ChaC glutathione-specific γ-glutamylcyclotransferase 1), which inhibits apoptosis in association with glutathione degradation and mitigation of endoplasmic reticulum (ER) stress response and inflammatory responses dependent on NFκB [48] was upregulated. This occurred in parallel with PSAT1 (phosphoserine aminotransferase 1), a protein involved in a glycolytic shunt pathway generating NADPH, which is required to maintain glutathione in a reduced form, facilitating mitochondrial antioxidant production [49].
Concurrent with these anti-caspase, anti-apoptotic responses, additional transcripts among those top 20 upregulated in narsoplimab-treated cultures encode proteins that limit inflammatory responses and/or promote vasculogenesis. They include: TM4SF18, a positive feedback modulator of vascular endothelial cell growth factor (VEGF) signaling and angiogenesis and a master regulator of the timing and magnitude of EC transcriptional signature decisions [50]; ASPM (abnormal spindle-like microcephaly-associated protein), a cell cycle gene linked to vascular regeneration and repair [51]; and ESM1 (endothelial cell-specific molecule 1), which promotes angiogenesis and forms a positive feedback loop with VEGF [52].
TNSF15 was the only member in the list of upregulated protein-encoding transcripts that did not reflect these salutary changes. This TNF superfamily member activates NFƙB and can induce EC apoptosis [53].
Narsoplimab-associated downregulation of transcripts for functional proteins
ZNF521 (zinc finger protein 521) is involved in the regulation of cell proliferation, decreasing Runx2 transcription and Akt phosphorylation leading to promotion of apoptosis [54].
Interleukin-1 receptor protein 1 (IL1R1) is involved in EC activation and injury in an autocrine and paracrine manner. Most interleukins acting on MVEC belong to the IL-1 cytokine group, increasing adhesion molecule expression, permeability, and inflammation and facilitating apoptosis [55].
Fibulin-5 is an extracellular matrix (ECM) glycoprotein that plays a critical role in vasculogenesis by suppressing angiogenesis factor angiopoietin/Tie-2 receptor- and VEGF-based signaling and enhancing expression of thrombospondin-1 in EC [56, 57]. This is of particular interest as angiopoietin-2 was one of the three biomarkers of endothelial injury correlating with TA-TMA development in a recent pediatric cohort [58].
Aggrecan, a major ECM proteoglycan that promotes apoptosis in vascular smooth muscle [59], was downregulated along with transcripts for SLC14A1. SLC14A1 is a primary transporter for urea and contributes to suppression of endothelial nitric oxide synthetase (eNOS) [60], and eNOS deficiency elevates aggrecan expression [59].
Lectin-like ox-LDL receptor-1 (LOX-1) is involved in promotion of EC apoptosis induced by oxidized LDL through the activation of ER stress sensors [61].
Transmembrane protein 204 (TMEM204, also known as claudin-like protein of 24 kd (CLP24)), is a hypoxia-regulated intercellular junction protein. TMEM204 suppression is associated with increased cellular adhesion and decreased cellular permeability [62].
Four among the top 20 down-regulated transcripts encode proteins the suppression of which could exacerbate EC apoptosis (BICD1 [63], DOCK9 [64], JUP (also known as plakoglobin) [65], and RAB11FIP2 [66]).
Discussion
We document the ability of the specific MASP2 inhibitor narsoplimab to suppress caspase activation induced in MVEC by plasmas from individuals with alloHSCT-associated TMAs, the majority of whom had high-risk disease. Utilizing RNAseq we identified a plausible mechanism by which narsoplimab could protect MVEC against TA-TMA-associated activation and injury. A synthesis of interactions among complement components, vascular growth factors, pro-inflammatory cytokines, and their signaling pathways, based on data from our in vitro model and earlier studies on known effects of MASP2 on complement and coagulation cascades, summarized in Fig. 5A and B, likely will define the ultimate outcome of narsoplimab administration in acute TA-TMA patients.
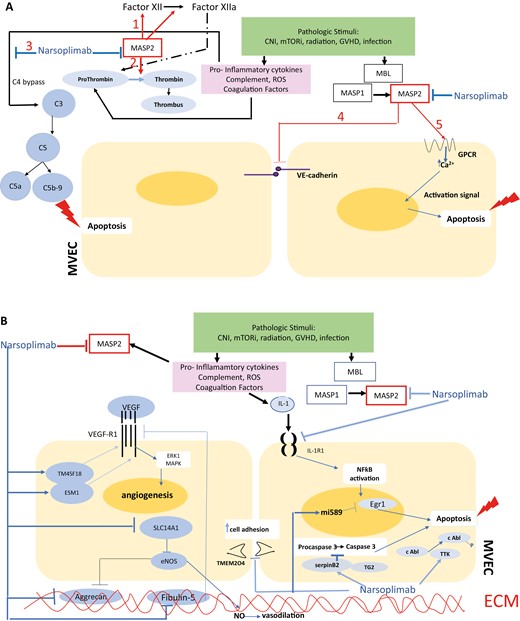
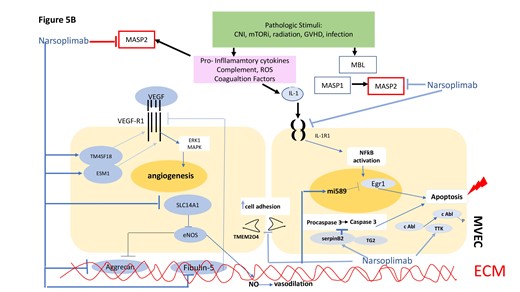
Graphic model of TA-TMA pathogenesis in terms of potential sites of intervention with the anti-MASP2 mAb narsoplimab. Proposed model for interacting cascades of complement, coagulation, pro-inflammatory and pro-apoptotic responses to microvascular endothelial cell injury leading to a TMA in the setting of an alloHSCT. Putative points of intervention by narsoplimab are indicated. A. Pathways for intervention with narsoplimab based on MASP2 interactions documented in the literature. As detailed in the text, a variety of EC modeling systems show that MASP2 can: amplify coagulation cascades via factor XII and prothrombin (Pathways 1 and 2); amplify the alterative pathway of complement through C4 bypass, leading to pro-inflammatory (C5a-based) conditions and EC apoptosis (C5b-9-based) (Pathway 3); facilitate endothelial permeability via disruption of VE-cadherin (Pathway 4); and elevate intracellular calcium-based signaling via GPCR, with resultant apoptotic signals (Pathway 5). B. Pathways for intervention with narsoplimab based on our RNAseq data.
The endotheliopathy characterizing TA-TMA in vivo is complex, with the potential for divergent pathways assuming dominant roles at different stages of the disorder. Apart from complement, adhesion molecules, angiopoietins, coagulation factors, pro-inflammatory cytokines, glycocalyx, and other ECM components, NETosis, NO, ROS, thrombomodulin, tight junction components, and toll-like receptors have all been implicated [58, 67]. These factors can be viewed in the context of three major stages in the pathophysiology of a TA-TMA [68]: an initiation phase comprising allotransplant conditioning regimens, including CNI, mTORi and radiation, acute GVHD, and infection, all leading to initial EC injury; a progression phase by which endothelial damage is aggravated by inhibitors of NO, VEGF, angiopoietins, and anti-apoptotic factors; and an outcome phase, including platelet aggregation over injured endothelium and positive feedback loops between coagulation and complement cascades. Based on its known activities in vivo, supported by data derived from our current RNAseq analyses, MASP2 has the potential to initiate and/or exacerbate all three stages, highlighting the rationale for intervention with a MASP2 inhibitor in TA-TMA. Indeed, the potential added value of a MASP2 inhibitor over anti-C5 agents or other complement inhibitors currently in clinical trials or in development for TA-TMA is the ability of such an agent to suppress components of both complement and coagulation cascades.
Specifically, the LP is activated by recognition of DAMPs exposed on injured endothelium by pattern recognition molecules, regardless of the mechanism(s) of that initial insult. They include MBL, ficolins, and collectin-11, which circulate in complex with MASP1 and -2. Upon sequestration to DAMP-presenting surfaces, MASP1 autoactivates and then cleaves MASP2, with subsequent cleavage by MASP2 of complement factors C2 and C4, leading to eventual formation of an inflammatory component, the anaphylatoxin C5a, and C5b-9, which can directly induce MVEC activation and injury [69]. In the presence of a MASP2 inhibitor such as narsoplimab, the LP ceases without formation of the C3 convertase complex, which is a prerequisite for the effector functions of the complement system [70]. Although not among the top 20 downregulated transcripts, it is noteworthy that narsoplimab suppressed transcripts for many complement proteins (Fig. 4B). Vascular EC, both microvascular and large vessel, expresses and releases complement proteins [71, 72], with the magnitude of such release by MVEC varying by tissue lineage [72].
There is also extensive crosstalk between the LP and coagulation, fibrinolysis, and kinin-kallikrein pathways. MASP2, a functional nexus between the complement and coagulation systems, can influence these interactions from multiple angles via cleaving prothrombin to generate thrombin as well as activation of clotting factor XII [73]; direct activation of EC, inducing an intracellular Ca2+ mobilization response, a classical sign of G-protein coupled receptor-mediated EC activation [70]; and disintegration of cell adhesion molecules, including VE-cadherin, markedly increasing endothelial permeability [65, 70]. These effects of MASP2 on EC behavior are thought to be of substantial physiological relevance as this protease is in direct contact with endothelium [70].
Although our work involved an isolated MVEC activation/injury model, the mechanisms by which MASP2 blockade could disrupt many of the pathway interactions outlined above are well-supported by RNAseq data derived from our system. Markers of complement activation, including elevated plasma levels and tissue deposition of C5b-9 and MASP2, inflammatory cytokine production (TNF-α, IL-1, IL-6, IL-8, IFN-ɣ), and endotheliopathy related to caspase activation and apoptosis all correlate with development and outcome of TA-TMA [2, 4, 5, 8, 10, 12]. Many of their effects should be mitigated by the narsoplimab-associated changes in transcripts for miRNAs and protein-coding genes illustrated here. Recent data also relate to the potential for narsoplimab to mitigate EC injury induced by immunosuppressive agents used in alloHSCT, which themselves may initiate a TMA. Such endotheliopathy appears to be both drug-class and medication specific. In terms of the CNIs, cyclosporine directly induces apoptosis in EC via caspase activation [74] suggesting, based on our data, that this should be blocked by narsoplimab. Tacrolimus also has direct EC toxicity but does not activate caspases [75], so narsoplimab may not suppress its effect. The mTORi everolimus does not induce caspase or directly injure EC [76, 77]. However, it can synergize with other treatment modalities, such as radiotherapy, to activate caspases and injure EC [77]. The mTORi sirolimus alone also fails to activate caspase, but it can induce EC autophagy [78]. Mycophenolate does not influence EC survival or caspase activation [75], supporting its use in place of a CNI or mTORi in the setting of TA-TMA.
Limitations to this study include the fact that transcript alterations determined by RNAseq were not confirmed utilizing individual RT-PCR analyses. However, our data are based on three replicate experiments suggesting that these changes are indeed reflective of the model. The concentrations of narsoplimab used were some 5-fold higher than required to suppress MVEC caspase activation induced by plasmas from patients with TTP and aHUS types of TMA in vitro, and higher than circulating levels reported in healthy volunteers and non-human primates [17, 33]. However, tissue levels of narsoplimab are likely to be much higher than plasma concentrations, and the circulating levels that are observed in vivo in humans are sufficient to induce clinical response in TA-TMA patients [17]. Multiple confounding factors apart from complement activation can contribute to TA-TMA development, including infection, inflammation, GVHD, and immune dysregulation [2], not all of which would be expected to be modeled in our MVEC system and/or affected by MASP2 blockade. For example, 2 among the top 20 upregulated transcripts and 4 among the top 20 down-regulated transcripts in narsoplimab-treated cultures implicate changes in miRNAs or protein-encoding genes which could exacerbate EC apoptosis. In addition, one of 9 TA-TMA clinical responders to narsoplimab whose plasma did not show a decrease in the ability to activate MVEC caspase 8 post-treatment had an ongoing cytomegalovirus infection and was on foscarnet therapy, either of which could have exacerbated MVEC injury. Finally, while pre-treatment plasmas from 8 of 9 narsoplimab clinical responders activated caspase 8 in MVEC pre-treatment, none of 4 clinical non-responders showed such activity, another indication that subsets of TA-TMA patients may have additional pathophysiologies.
Despite these confounding factors our data, viewed in the context of the recent clinical trial of narsoplimab in high-risk TA-TMA [17], support the use of this drug in TA-TMAs, as well as consideration for its investigation as a prophylactic measure in high-risk alloHSCT patients, and perhaps expansion to other forms of complement-associated endotheliopathy. For example, the considerable interaction between caspase-dependent cell death pathways and cytokine signaling has raised the potential of caspase 8 as a target in suppression of progression of microthrombotic disorders such as COVID-19 [79, 80].
Abbreviations
- alloHSCT
allogeneic hematopoietic stem cell transplant
- aHUS
atypical hemolytic uremic syndrome
- CNI
calcineurin inhibitor
- mAb
monoclonal antibody
- GVHD
graft versus host disease
- MBL
mannose-binding lectin
- MASP2
mannose-binding lectin associated serine protease 2
- MAC
membrane attack complex
- miRNA
microRNA
- mTORi
mammalian target of rapamycin inhibitor
- MVEC
microvascular endothelial cell
- RPKM
Reads Per Kilobase of transcript per Million mapped reads
- RNAseq
RNA sequencing
- ROS
reactive oxygen species
- TA-TMA
transplant-associated thrombotic microangiopathy
- TTP
thrombotic thrombocytopenic purpura.
Acknowledgements
Not applicable.
Ethical Approval
Approved by the WCM Institutional Review Board, registered with the National Clinical Trials Network (NCT02604420), and informed consent was obtained from all subjects.
Conflict of Interests
JL has received grants from Omeros Corporation, manufacturer of narsoplimab, and grants and honoraria from Alexion, Inc. and Jazz Pharmaceuticals. The remaining authors declare no competing financial interests.
Funding
This study was supported by grants from Omeros Corporation and the Angelo Donghia Foundation (JL).
Data Availability
The RNAseq data are available by contacting Jeffrey Laurence: [email protected].
Author Contributions
All authors were involved in the analysis, and interpretation of data and approved the final version of the manuscript. JL: concept and design and drafting of the manuscript. SE and JL compiled clinical data from record reviews. SE performed the ELISA assays and RNA isolations. AT performed the RNAseq assays. DR, AT, JH, SE, and JL analyzed the RNAseq data. JH and DR performed statistical analyses.
Permission to Reproduce
Not applicable

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}