





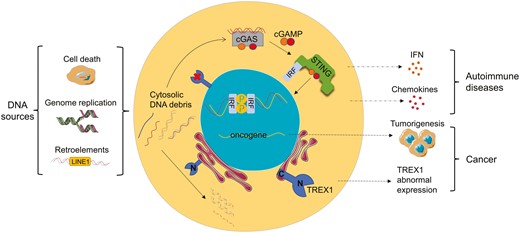
Introduction
Three prime repair exonuclease 1 (TREX1) is one of the crucial exonucleases in mammalian cells. It mainly anchors on the cytoplasmic side of the endoplasmic reticulum (ER) membrane and plays the role of 3ʹ-5ʹ exonuclease as a homodimer, which degrades aberrantly accumulated DNA debris in the cytoplasm, preventing it from triggering autoimmunity [1]. It is also known as deoxyribonuclease III (DNase III) and is a member of the DEDD exonuclease family, which is defined as having a conserved Asp-Glu-Asp-Asp (DEDD) motif [2, 3]. TREX1 shows a similar catalytic ability to other enzymes in the same family, but it is the only one that degrades double-stranded DNA (dsDNA) [4]. Notably, TREX1 deficiency or depletion results in aberrant DNA accumulation in the cytosol, including single-stranded DNA (ssDNA) and/or dsDNA fragments [5]. As the front line of defense in innate immunity, pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) and generate initial immune responses. DNA is the major genetic material of pathogens and can be recognized as PAMPs by Toll-like receptors (TLRs) or DNA sensors [6, 7]. For instance, TLRs recognize DNA substrate and activate IFN regulatory factor (IRF) or nuclear factor kappa B (NF-κB), resulting in the production of interferons and pro-inflammatory factors [8]; AIM2-like receptors (ALRs), such as IFN-inducible 16 (IFI16), function as intracellular DNA sensors to recognize dsDNA and elicit interferon responses [9]. Cytosolic self-DNA debris will also be recognized as a damage-associated molecular pattern (DAMP), triggering inflammation or specific immunity [10].
The major DNA sensor in mammalian cells is cyclic GMP-AMP synthase (cGAS), which is mostly localized in the nucleus and tightly bound to nucleosomes with very low activity or in an inactive form [11]. To avoid inappropriate self-DNA detection, cGAS localizes to the plasma membrane in the resting state [12]. However, the cytosolic cGAS directly binds dsDNA and catalyzes the synthesis of the cyclic GMP-AMP (cGAMP), which stimulates the production of IFN-I and inflammatory factors through stimulator of interferon genes (STING) signal pathways, such as STING-TBK1-IRF3 and STING-IKK-NFκB [13]. Under the activation of dsDNA, cGAMP can also be transferred to adjacent cells through gap junctions to activate their STING pathways [14], which further amplifies the immune response. Thus, the inflammatory signal cascades caused by TREX1 malfunction trigger a wide range of tissue inflammation and/or autoimmune diseases. For example, the absence of TREX1 in mice resulted in autoimmune phenotypes such as inflammatory myocarditis [15]. Also, Aicardi-Goutières syndrome (AGS), systemic lupus erythematosus (SLE), retinal vasculopathy with cerebral leukodystrophy (RVCL), and familial chilblain lupus (FCL) have been observed in TREX1-deficient patients [16, 17]. Interestingly, removing even one cGAS allele improves the phenotypes in TREX1-knockdown mice, and complete elimination of cGAS eradicates all inflammation and pathological manifestations [18, 19]. However, the presence or absence of ALRs does not change the phenotypes [20]. Apparently, cGAS-STING pathway is prominently engaged in the self-DNA immune response, and other inflammatory pathways may have also been involved in TREX1-related autoimmunity due to the presence of various proinflammatory cytokines in patients [21].
Here, we explore the origin of cytosolic DNA associated with TREX1 deficiency, analyze the mechanisms by which TREX1 degrades DNA, and highlight the connections between TREX1 and diseases. It is hoped that these contents will lead to the discovery of new research avenues and therapeutic solutions.
The structure and function of TREX1
Given the previous understanding of protein structure based on mouse TREX1 (mTREX1) [22–27] and human TREX1 (hTREX1) [28], the spatial structure of TREX1 is essentially definite, but there are some differences in amino acid composition and related functions between mTREX1 and hTREX1. Encoded by a single exon gene located at chromosome 3p21.31, TREX1 exists as a stable protein homodimer. The main chains of two protomers contact each other, forming a stable and central antiparallel β fold that extends the dimer’s length, and extensive hydrogen bonds are found between the side chains and the main chains. One protomer is composed of 314 amino acids and the 242 amino acids of the N-terminal contributes to catalytic activity [23, 29] (Fig. 1A). In each active site, Asp-18, Glu-20, Asp-130, and Asp-200 coordinate with two divalent metal ions [23] to fully catalyze the polynucleotide substrates [24, 25]. Mutations at the three residues, Asp-18, Asp-200, and His-195, were shown to affect the coordination between the DEDD motif and metal ions and impair the catalytic activity [22]. In a homodimer, two enzymatic pockets are located on the opposite outer edges of the dimer interface. The residues contact 18 water molecules in the core of the pocket [28], which provides potential drug combinations. Besides, several key residues in the N-terminal play roles in protein stability. Pro-10 directly contacts Asp-220 near the C-terminal to form a salt bridge, which stabilizes the formation of protein pockets and crystallization. In primate TREX1, two determinant residues, Phe-17 and Met-19, are highly conserved, and mutations at either site affect the protein’s thermal stability. While in low-level mammals, these two positions are Leu-17 and Leu-19 [28]. Arg-62 of one protomer communicates across the dimer interface to promote the catalysis of the opposing protomer, ensuring the structural integrity of the dimer [30]. Arg-97 also crosses the interface to form hydrogen bonds with Asp-65 and Gln-115, and Arg-114 directly connects with Cys-99 and Gln-98 in the opposite to stabilize protein dimerization [28] (Fig. 1B). Studying the roles and interactions of these determinant residues in the N-terminal helps us better comprehend the mechanisms underlying TREX1 mutations in autoimmune diseases. Furthermore, the residues and functional differences between mTREX1 and hTREX1 imply that the same targeted drug may have different efficacy in treating animals and humans.
![The crystal structure and critical residues of the hTREX1 dimer. A monomer only contains 242 amino acids of the TREX1 catalytic domain, and the cartoon is visualized using Protein Data Bank (PDB) structure 7TQQ from Ref [28]. (A) hTREX1-DNA complex. The color of the protein homodimer is distinguished by the secondary structure, with β-folds being yellow; (B) Key residues of TREX1. The enzymatic pockets are highlighted by circular dashed regions. Pro-10 directly contacts Asp-220 to form a salt bridge. Phe-26, Arg-128, Lys-160, and Arg-164 contact the 5ʹ-end of DNA. Three rectangular dashed regions: (a) The active site of the pocket. Asp-18, Glu-20, Asp-130, and Asp-200 directly coordinate with two divalent metal ions, and His-195 is involved; (b) Arg-62 of one protomer gets across the dimer interface into the opposing protomer. Arg-97 crosses the interface to form hydrogen bonds with Asp-65 and Gln-115 in the opposite, and Arg-114 connects with Cys-99 and Gln-98 in the opposite; (c) 10 residues (Asp-18, Glu-20, Ala-21, His-124, Asp-130, Ser-176, Tyr-177, Leu-179, His-195, and Asp-200) are in contact with the 3ʹ-end of DNA](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cei/211/3/10.1093_cei_uxad017/2/m_uxad017_fig1.jpeg?Expires=1747862376&Signature=xlY1knZRNyaUwzm5Jlh-576xf0gtJ42AiG2POahI3d5hGKUkkapb1bDoFKE2IvVvn7rdnkAD~MUPW7E1FPgs6w36xG0yCoIktJZ9q1BoorGuJQ4SxUD2wJ6Tv9AB5SX4kgFG74Rm4UVGPrOZ1Fa8LkirtE2BZEvasfjh7-7FFM5y3ENqurtwUx194pgaI0xbtc2mxg~kngUoGitDM5mAJyc2arEaNkHJDaMKyfy8JeGOYc~57dUIS3rsEe9lKjdf1Pq27y6VXyVjBs0jZWSmm0Fp-2ubol3B-avJEMy4O9gaa1i2vnTtYy2ojfJAS8EXa1socvy84rSwFUDMW2nWiA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The crystal structure and critical residues of the hTREX1 dimer. A monomer only contains 242 amino acids of the TREX1 catalytic domain, and the cartoon is visualized using Protein Data Bank (PDB) structure 7TQQ from Ref [28]. (A) hTREX1-DNA complex. The color of the protein homodimer is distinguished by the secondary structure, with β-folds being yellow; (B) Key residues of TREX1. The enzymatic pockets are highlighted by circular dashed regions. Pro-10 directly contacts Asp-220 to form a salt bridge. Phe-26, Arg-128, Lys-160, and Arg-164 contact the 5ʹ-end of DNA. Three rectangular dashed regions: (a) The active site of the pocket. Asp-18, Glu-20, Asp-130, and Asp-200 directly coordinate with two divalent metal ions, and His-195 is involved; (b) Arg-62 of one protomer gets across the dimer interface into the opposing protomer. Arg-97 crosses the interface to form hydrogen bonds with Asp-65 and Gln-115 in the opposite, and Arg-114 connects with Cys-99 and Gln-98 in the opposite; (c) 10 residues (Asp-18, Glu-20, Ala-21, His-124, Asp-130, Ser-176, Tyr-177, Leu-179, His-195, and Asp-200) are in contact with the 3ʹ-end of DNA
The 72 amino acids at the C-terminal are mostly related to cellular localization [31], with the last 30 amino acids being highly conserved and hydrophobic, forming a transmembrane helical structure. Since the TREX1 gene lacks an identifiable membrane insert signal sequence, the protein anchors on the ER membrane and the outer membrane of the nuclear membrane or interacts with ER-resident proteins to determine its cellular localization [32, 33]. Moreover, the highly conserved sequence at the C-terminal controls the ubiquitination of TREX1 to regulate cellular localization. It interacts with ubiquilin 1 and recruits TREX1 and cytosolic DNA into vesicles generated during autophagy to further degrade DNA [34]. Therefore, mutations at the C-terminal that contribute to aberrant localization can also result in abnormal DNA accumulation in the cytosol and trigger immune responses. In addition, the C-terminal interacts with oligosaccharide transferase (OST) across the ER membrane, co-glycosylates lipid-linked oligosaccharide (LLO), and affects the OST activity. Loss of glycosylation function allows immunogenic substances, such as erroneously hydrolyzed LLO and many free oligosaccharides, to accumulate in the cytosol, triggering autoimmune responses through the STING-TBK1-IRF pathway [35, 36] (Fig. 2). To avoid immune activation without affecting enzymatic activity, TREX1 phosphorylation in the C-terminal inhibits its interaction with the OST complex during karyomitosis [37]. Under cell stress or cell division in the S phase [5], living cells quickly repair their ruptured nuclear membrane, but aberrant nuclear compartments (which are called micronuclei) wrapping micronuclear DNA remain. The tether between TREX1 and ER helps TREX1 localize to ruptured micronuclei and then exert enzymatic activity [38]. TREX1 also exists in ER-linked macromolecular complexes with other endonucleases to synergistically degrade DNA produced in granzyme A-mediated cell death [39, 40]. Furthermore, TREX1 activities have been found to be associated with chromatin in the nucleus and to participate in DNA damage repair via Poly (ADP-ribose) Polymerase-1 [41]. Further studies on the precise locating capability of the last 30 amino acids should be brought forward for drug orientation.
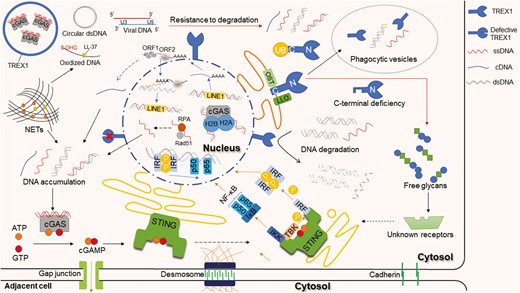
TREX1 functions and the aberrant DNAs in the cytosol trigger signal pathways. The N-terminal of TREX1 is responsible for degrading DNA, including ssDNA and dsDNA. The C-terminal engages in protein localization and can be regulated by ubiquitination. It interacts with ubiquitin (UB) and recruits TREX1 and cytosolic DNA into vesicles. The C-terminal also interacts with OST and co-glycosylates the LLO. The cytosolic DNA is mainly derived from abnormal DNA replication in the nucleus, cell death, and endogenous retroelements. For example, ssDNA-binding proteins RPA and Rad51 deficiency lead to the accumulation of ssDNA; NETs released from neutrophils contain DNA scaffolds that come from the nucleus and/or mitochondria; non-LTR retroelement LINE-1 retrotransposition produces ssDNA. The DNA sensor cGAS directly binds cytosolic dsDNA and catalyzes the synthesis of cGAMP, which then stimulates the downstream STING-TBK1-IRF3 and STING-IKK-NFκB pathways. cGAMP can transfer to adjacent cells through gap junctions. There are some mechanisms of resistance to TREX1, including cGAS-DNA phase separation, DNA oxidation, circular DNA, and HIV DNA formation
The sources of aberrant DNA in the cytosol
In mammalian cells, dsDNA is strictly confined to the nucleus and mitochondria. The genetic information is transcribed into mRNA that traverses the nuclear pore into the cytoplasm. During the interphase S of cell division, dsDNA forms replication forks with the assistance of DNA-binding proteins, helicases, polymerases, and other proteins, and completes DNA semiconservative replication. Given the abundance of repetitive DNA sequences in the human genome, DNA fragments may unintentionally enter the cytosol as a result of replication program errors. It has been demonstrated that DNA replication is an essential source of DNA waste for triggering inflammatory responses [42]. For example, the obstructive formation of the replication fork and malfunctions in the synthesis and decomposition of leading or lagging strands are potential sources of ssDNA [43]. During DNA replication and repair, two key proteins, the ssDNA-binding protein RPA (replication protein A) and Rad51 (recombination protein A), protect ssDNA from degrading during elongation, prevent ssDNA reannealing, and coordinate the assembly of processing factors. Normally, RPA and Rad51 confine ssDNA in the nucleus and prevent it from crossing the nuclear membrane. Their deficiency leads to the accumulation of ssDNA in the cytosol and triggers the IFN response via cGAS [33, 43]. Upon cellular stress, programmed cell death, necrosis, or the formation of neutrophil extracellular traps (NETs), the nuclear membrane ruptures and DNA/RNA debris is free in the cytosol [44] (Fig. 2). Different from apoptosis and necrosis, NETs also function as immune barriers and are formed in two pathways. One is NETosis in a slow death mode, which involves neutrophil chromatin decondensation and plasma membrane rupture to release NETs. The other is suicidal NETosis, which involves the rapid expulsion of nuclear chromatin and granule proteins to form NETs in the extracellular space [45]. The NETs, comprising antimicrobial peptides and DNA scaffolds that come from the nucleus and/or mitochondria, wrap and kill pathogens, and are finally phagocytosed by macrophages [46]. So far as it is known, the degradation of NETs mainly depends on DNase I and TREX1, whose dysregulation also contributes to DNA accumulation and immune diseases [47, 48].
However, TREX1 may be an unwitting accomplice to some pathogens. For instance, the human immunodeficiency virus (HIV) invades immune cells, and the viral RNA reverse transcribes into cDNA, which enters the nucleus to synthesize dsDNA. The viral integrase removes the 3ʹ-end GT dinucleotide of dsDNA and inserts the recessed 3ʹ-end into the opposite strand of the host chromosome to propel gene integration [49]. Abortive cDNA that fails to enter the nucleus, as well as unprocessed dsDNA, will be destroyed by TREX1, preventing them from triggering innate immunity in the cytoplasm. Therefore, HIV cDNA could accumulate and activate IFN-related immune responses in TREX1-deficient cells [50]. Similar to HIV, endogenous retroviruses (ERVs) and endogenous retroelements may also be sources of DNA in the cytosol [9, 51]. ERVs and retroelements are part of the DNA left in the human genome after the ancient retrovirus infected the germline. Most of them are in a state of silence, and only a few pieces of DNA can be produced under the action of reverse transcriptase. There are two types of retroelements. One is the long terminal repeat (LTR), which still retains the gene structure for simple virus synthesis. The other is non-LTR, which lacks the genes for synthesizing capsid and envelope proteins and only conducts reverse transposition in the nucleus to produce DNA fragments [52]. Long interspersed nuclear elements-1 (LINE-1) is a rich non-LTR retroelement in the mammalian cell genome [53] and it is highly active in the nervous system. In TREX1-defective nerve cells, ssDNA produced by LINE-1 retrotransposition accumulates in the cytosol, and then becomes neurotoxic and triggers an IFN response, which possibly contributes to the neuropsychiatric symptoms in human autoimmune diseases [54, 55] (Fig. 2). Nonetheless, the DNA generated during LINE-1 retrotransposition is not exclusive to the initiation of autoimmunity. It can also stimulate IFN-β production via RNA pathways [56]. In addition, studies have shown that TREX1 deficiency does not increase LINE-1 retrotransposition in mice, whereas SAM and HD domain-containing protein 1 (SAMHD1) and RNaseH2 deficient mice have significantly increased LINE-1 retrotransposition [57, 58]. The regulation of LINE-1 retrotransposition seems different between humans and other mammals. It may be co-regulated by a variety of enzymes, and TREX1 is only one of them.
Degrading mechanisms
TREX1 primarily binds to the aberrant DNA debris accumulated in the cytosol and then degrades them into deoxynucleotide monophosphates. In general, TREX1 is in close contact with both DNA strands, with 10 residues (Asp-18, Glu-20, Ala-21, His-124, Asp-130, Ser-176, Tyr-177, Leu-179, His-195, and Asp-200) in contact with the 3ʹ end and 4 residues (Phe-26, Arg-128, Lys-160, and Arg-164) in contact with the 5ʹ end. Lys-160 and Arg-164 make direct contact with the DNA phosphate backbone to stabilize the 5ʹ end, and Arg-164 is present only in human and primate TREX1 sequences [25, 26, 28]. Arg-128 destabilizes dsDNA and presents ssDNA to the catalytic site, enhancing the efficiency of dsDNA degradation [59, 60].
Specifically, the bonds have two conformational forms within the active site. One is “tight conformation”: TREX1 active site applies a base-flipping mechanism to bind and unwind DNA double strands, thereby degrading polynucleotides more efficiently. The other is “loose conformation”: TREX1 binds directly to a single strand of DNA that needs to be degraded [25]. If a base at the 3ʹ-end of dsDNA is damaged by irradiation or oxidation, the hydrogen bonds between the base pair fail to form. TREX1 will insert the leu24-Pro25-Ser26 cluster into this unstable double helix structure, using a catalytic active site to cut the damaged end (Fig. 3A). While redundant nucleotides appear at the 3ʹ-end of dsDNA, TREX1 will cap the non-scissile 5ʹ-end with the leu24-Pro25-Ser26 cluster, and then cut off the 3ʹ-overhang (Fig. 3B) [26]. Considering that TREX1 and the DEDD family member RNase T from Escherichia coli, perform similar catalytic activities, both share the same base preference and are able to cut deaminated bases in ssDNA like uracil and hypoxanthine. TREX1 acts 3–4 times more efficiently on ssDNA degradation compared to that of dsDNA [5]. The aberrant ssDNA inserts its 3ʹ-end into the enzymatic pocket of the TREX1 dimer, and the last nucleotide will be fixed by Leu-24 and Ile-84 in the active site (Fig. 3C). With the assistance of Mg2+, TREX1 completes the hydrolysis of the phosphodiester bonds [26]. Meanwhile, TREX1 can directly degrade ssDNA generated by reverse transcription of retroelements [32] or inhibit the production of LINE-1 cDNA by clearing open reading frame 1 of LINE1 with the assistance of proteasomes [61].
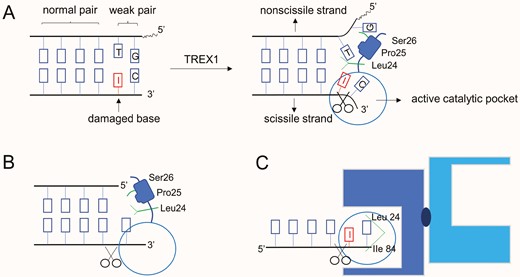
Patterns of DNA degradation by TREX1. (A) An adenine at the 3ʹ-end of dsDNA is damaged by irradiation; TREX1 disassembles the wake pair and cuts the damaged end; (B) TREX1 cuts off the redundant nucleotides at 3ʹ-end of dsDNA; (C) ssDNA inserts its aberrant 3ʹ-end into the catalytic domain of TREX1
TREX1 also plays a role in DNA repair, including cleaving single-stranded fragments with mismatched ends in the DNA double strands [26]. However, in out-of-control cancer cells, TREX1 may cause secondary damage to DNA in the nucleus with other endonucleases, and even drive the transition of cancer cells from in situ to invasion [62]. Surprisingly, studies showed that the catalytic sites of TREX1 dimer could also bind RNA and degrade ssRNA, RNA–DNA complexes, and tRNA; though the efficiency was lower than that of DNA degradation. TREX1 was also found to interact with the endonuclease RNase H2 to jointly complete the effective removal of RNA fragments. Besides, there are excessive loads of RNA–DNA hybrids in AGS patients’ fibroblasts due to the weakened DNA methylation capacity [63, 64]. The specific mechanism of RNA degradation by TREX1 and its role in autoimmune diseases remain to be further investigated.
Apart from the widely known nuclease activity, TREX1 forms phase-separated droplets with cGAS-DNA to resist TREX1-mediated DNA degradation [65]. TREX1 localizes exclusively to the outer periphery of the droplets with weak catalytic activity and forms a “shell” around the inner compartment, which contains cGAS-DNA condensates, impeding the likely immune sensing. Moreover, TREX1 E198K shows an impaired ability to degrade the phase-separated DNA, indicating that Glu-198 has a special function in phase separation formation [27]. In addition to physical repulsion, structurally altered DNA also exhibits resistance to TREX1-related DNA degradation. For example, when circular plasmid DNA was incubated with TREX1, the nicked polynucleotide strand was nearly completely degraded, while the intact ssDNA strand was retained [59]. It demonstrated that a circular, un-nicked DNA would be resistant to the degradation. In addition, oxidized DNA that occurs during the formation of antimicrobial reactive oxygen, in the NETs during the oxidative burst, or in response to certain physical damage such as the absorption of UV light, also shows resistance. The antibacterial peptide LL-37, in conjunction with the oxidized base 8-hydroxyguanosine (8-OHG) containing DNA in NETs, was discovered to promote the immune response by reducing DNA sensitivity to degradation [66]. On one hand, LL-37 allows delivery of self-DNA into cytosol [66]; on the other hand, it stabilizes the DNA against degradation by bacterial nucleases [67]. So, it is reasonable to speculate that LL-37 may, to some extent, assist in the resistance to TREX1-related DNA degradation. Furthermore, it was found that before HIV integration, reverse transcribed viral dsDNA is processed to produce a 5ʹ-overhang and a recessed 3ʹ-end [49]. The 3ʹ-processing viral DNA substrates, either ssDNA or dsDNA, were resistant to degradation. However, non-viral DNA with the 3ʹ and 5ʹ ends modified can still be degraded by TREX1. For DNA substrates containing both modifications and complementary (U3 and U5) viral sequences, TREX1 resistance will be present [68]. Therefore, U3 and U5 sequences of HIV DNA may have unique features such as secondary structures that work together with the modifications to block the active sites of TREX1 (Fig. 2).
TREX1 and autoimmune diseases
As mentioned above, TREX1 plays critical roles in cells, such as repairing DNA damage, degrading aberrant DNA fragments in the cytosol, and assisting OST in glycosylation. Apparently, DNA metabolism will be in a mess when TREX1 loses its capabilities. Studies have shown that TREX1-deficient cells exhibit G1/S phase transformation defects and chronic Ataxia Telangiectasia Mutated (ATM)-dependent checkpoint activation. The ssDNA would be continuously produced in the S phase and accumulate in the ER even without exogenous stress [69]. In TREX1 D18N and TREX1 D200N cells, the TREX1 homodimer binds to the dsDNA but cannot exert its catalytic activity, thus forming the DNA-TREX1 complex and inhibiting the degradation of DNA [25, 59]. It is demonstrated that the accumulation of 60–65 base ssDNA in the cytosol of TREX1-deficient cells is sufficient to trigger innate immune responses in mice and humans [5]. In animal models of TREX1 D18N mice, immune cells are abnormally activated to produce large amounts of IFN-I and anti-dsDNA antibodies, and then IFN-related systemic inflammatory phenotypes, including lymphadenectasis, vasculitis, and kidney disease are manifested [25, 70]. The mice with a complete absence of TREX1 develop inflammatory myocarditis [15, 18]. In humans, TREX1 gene defects are believed to be closely related to AGS [71, 72], SLE [73, 74], FCL [75, 76], and RVCL [77]. For mutations leading to AGS, SLE, and FCL, most of them are missense or nonsense at the N-terminal of TREX1, while they are frameshift at the C-terminal in RVCL [78] (Fig. 4).
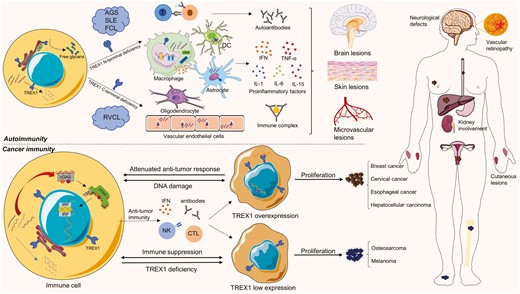
Pathogenesis of TREX1-related diseases. TREX1 deficiencies are associated with autoimmune diseases such as AGS, SLE, FCL, and RVCL. Most of AGS, SLE, and FCL are caused by N-terminal deficiencies of TREX1, while C-terminal deficiencies result in RVCL. Cells with deficient TREX1 primarily activate cGAS-STING pathways through cytosolic DNA accumulation and other immunogenic substances. The inflammatory products released by many kinds of immune cells, for example, dendritic cells (DCs), astrocytes, oligodendrocytes, macrophages, and endothelial cells, trigger a wide range of tissue inflammation in autoimmune diseases, especially the lesions in the brain, skin, and microvessels. TREX1-mutated autoimmune diseases mostly manifest neurological symptoms, lupus-like cutaneous lesions, and microvascular defects. In cancer immunity, TREX1 expression can be adaptably regulated to facilitate tumor proliferation. Since cancer cells have three powerful weapons to escape immune tracking, TREX1 overexpression is beneficial to prevent the immune response and may induce the expression of proto-oncogenes. Low TREX1 expression could be a sign of immune suppression and TREX1 deficiency. The corresponding tumor types are listed
AGS is a rare hereditary encephalopathy that is classified into nine types according to the mutated genes (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1, LSM11, and RNU7-1) [79]. The typical manifestations include microcephaly, intracranial calcification, white matter abnormalities, and increased IFN-α in cerebrospinal fluid. AGS patients usually manifest multiple nervous system injuries, muscular rigidity, and cognitive impairment. In addition, they also show other symptoms such as chilblains, lupus-like skin lesions, and other autoimmune characteristics [80]. A recent study showed that the TREX1 mutant (AGS1) accounted for 17% of AGS, and patients with AGS1 had the earliest onset of neurological abnormalities and the most severe phenotype among all types [81]. Astrocytes, which are widely distributed in the central nervous system, are the main source of IFN [82]. The accumulation of cytosolic DNA caused by TREX1 deficiency leads to an IFN response, and TREX1-deficient cells also rely on cGAS to activate more inflammatory response of surrounding cells through cell junctions [83]. Researchers found that inhibiting the TREX1 gene induces apoptosis in astrocytes and slows differentiation in endothelial cells. It shows that TREX1 not only plays a role in degrading DNA in astrocytes but also regulates cell growth and differentiation [84]. This chain reaction of immune inflammation may be one of the reasons for the early onset of abnormalities in AGS1 patients. SLE is a systemic, multi-etiological autoimmune disease. Abnormalities in genes, hormones, and environmental factors may contribute to its pathogenesis. Patients may have no typical signs in the early stage, and different people may have different clinical symptoms, such as lupus-like skin lesions, neurodevelopmental disorders, kidney involvement, etc., with mild or severe presentations. As far as the pathogenesis of SLE is concerned, the formation of autoantibodies, the deposition of immune complexes, and the activation of chemokines and interferon are mostly manifested [74]. Immunoregulatory impairments lead to the accumulation of anti-nuclear antibodies, in which anti-dsDNA antibodies serve as major diagnostic or prognostic indicators of SLE [85]. As a potential source of self-antigen, NETs may also contribute to the pathogenesis of SLE. Previous studies have shown that SLE patients failed to dismantle the NETs due to aberrant DNase I function in the serum [86]. NETs from SLE patients, unlike those from healthy people, stimulate dendritic cells (DCs) to produce IFN-α, which in turn stimulates neutrophils to release NETs, forming a positive feedback loop [87]. Statistics have shown that the TREX1 mutation is present in about 2% of SLE patients [17]. And the digestion of NETs by TREX1 in macrophages and by DNase1L3 in DCs sheds more light on the potential role of NETs in SLE with TREX1 dysfunction [48]. FCL is a monogenetic inherited cutaneous lupus, mostly inherited in autosomal dominant pattern. It is often characterized by vascular inflammatory lesions and the production of autoantibodies, which are believed to be related to TREX1, SAMHD1, and STING [88, 89]. AGS, FCL, and SLE share some phenotypes, such as lupus-like skin lesions, autoantibody production, increased IFN levels, and neurological symptoms, with a few TREX1 gene mutations appearing in them simultaneously. Chilblain lupus is described as a cutaneous form of SLE, and FCL can in some ways be considered as a non-neurological form of AGS [17]. Diseases that exhibit this range of symptoms are therefore often referred to as lupus-like autoimmune diseases and the severity is commonly related to the degree of TREX1 deficiency.
RVCL is a neurovascular syndrome caused by TREX1 C-terminal mutations. The lesions can be observed in the brain, retina, kidney, and other microvessels, resulting in abnormalities of the corresponding blood supply tissues [90]. Mutations in the C-terminal of TREX1 have also been found in simple renal thrombotic microangiopaplasia [91] or vascular retinopathy [92]. The microvascular lesion could be related to the abnormal localization of TREX1. It is normally anchored on the ER, but it is now free in the cytoplasm or appears in the nucleus, still being catalytic [93]. An autopsy of an RVCL patient showed that TREX1 protein was positive in the nucleus of vascular endothelial cells and oligodendrocytes in the lesion area, which further verified the pathogenesis discussed above [94]. Whether C-terminal abnormalities induce the unexpected entry of TREX1 into the nucleus affecting the composition and decomposition of DNA, trigger the cytosolic accumulation of free glycans producing the immune response, and impair the N-terminal enzymatic activity, the exact mechanism remains to be further studied.
Even for the same disease, patients with different gene mutations have different manifestations [95, 96]. Mutations at different gene loci of TREX1 lead to different amino acid variations, different enzymatic activities, as well as various levels of free self-DNA and cGAS in the cytosol, which may affect the initiation and regulation of signal pathways [13]. The superposition of immunogenic substances such as free glycans and self-nucleic acid may also aggravate the inflammatory response [97]. The accumulation of aberrant self-DNA caused by TREX1 deficiency may be an important etiology of related diseases, but it is not the only pathogenic factor. In-depth studies of the function and location of TREX1, the source and path of self-nucleic acid, and the proportion of different immunogenic substances in the formation of the disease are the key factors to unlock this kind of autoimmune disease. A deeper understanding of TREX1 may provide more options for prevention, treatment, and disease prognosis in the future.
TREX1 and cancer immunity
Cancer cells are transformed from cells with mutations in proto-oncogenes and tumor suppressor genes. Normally, the body would recognize and eliminate such cells immediately. For example, immune cells engulf or attack aberrant cells, and enzymes repair or degrade abnormal nucleic acids. Therefore, cancer cells have evolved three powerful weapons to escape tracking. First, prevent the immune system from recognizing its abnormal DNA; second, destroy IFN-related immune mechanisms and induce the expression of proto-oncogenes; third, establish an immune suppression system to defend against the anti-cancer response [98] (Fig. 4). TREX1 degrades aberrant DNA in unstable cells, preventing them from becoming cancerous. Otherwise, the rest of tumor-derived DNA activates the cGAS-STING pathway to promote IFN-dependent anti-tumor responses. Both intrinsic and extrinsic STING were found to be involved in anti-tumor immunity [99]. Cytosolic DNA directly activates the intrinsic STING pathway in tumor cells; while these tumor-derived DNA can also trigger extrinsic STING pathways in immune cells through efferocytosis [100].
Some tumor cells overexpress TREX1 to prevent the immune system from recognizing their DNA and initiating IFN-dependent immunity. In human cervical tissue samples, TREX1 levels were lower in the normal cervical epithelium but higher in precancerous lesions, squamous cell carcinomas, and adenocarcinoma specimens. Up-regulation of TREX1 expression is thought to promote the growth of cervical cancer cells [101]. In another study of breast cancer, high TREX1 expression was associated with a lower survival rate in patients. When cancer cells expand within the space-restricted mammary ducts, they are easily squeezed and ruptured. Then TREX1 moves from the ER to the nucleus, causing chronic DNA damage. Loss of the ATM-dependent senescence-associated proliferation arrest activates the metastasis of cancer cells in situ [62]. It provides another possibility of direct DNA damage caused by high expression of TREX1 during tumorigenesis. In addition, TREX1 levels are also upregulated in hepatocellular carcinoma [102] and esophageal squamous cell carcinoma. TREX1 was found to be mainly distributed in the nucleus of esophageal cancer cells, leaving less TREX1 on the cell membrane than that in adjacent tissue cells [103]. Together, these studies highlight that elevated TREX1 expression in different cancer cells may serve to reduce immunogenicity or increase the vicious cycle of DNA damage. Therefore, inhibition of TREX1 and enhancement of the cGAS-STING pathway-mediated interferon response have become the keys to anti-tumor therapy.
In a radiotherapy study on cancer cells, single doses of more than 12–18 Gy could induce TREX1 to degrade DNA in the cytosol, reducing immunogenicity and inhibiting IFN secretion, which is counterproductive to immunotherapy. Accordingly, repeated irradiation with a radiation dose below the threshold of inducing TREX1 expression may be a better choice. Such a method better stimulates IFN production and initiates CD8+ T cells by activating DCs. Further combining it with immune checkpoint inhibitors at the same time can reactivate immune cells and eliminate cancer cells [98]. Notably, methods that directly target TREX1 also work due to the increased understanding of TREX1 protein structure and gene sequence. For instance, down-regulation of TREX1 gene expression improves the immunogenicity of tumor cells, driving inflammatory factors into the tumor microenvironment, and inhibiting tumor growth through immune activation and induction of Fas and TRAIL receptor expression [104]. In cervical cancer cells containing high-risk HPV protein expression, TREX1 gene silencing also inhibits tumor cell growth by inducing P53 upregulation and subG1 cell accumulation [101]. When treated by the DNA damage response inhibitor ceralasertib, depletion of TREX1 in THP-1 tumor cells also increases type I interferon expression and DC activation by transactivating the STING pathway [100]. Midostaurin, a multiple kinase inhibitor, could downregulate TREX1 protein expression with an increase in cytosolic DNA and augment the mRNA expression of cGAS, STING, and IRF3, which inhibit colorectal adenocarcinoma cell growth [105]. Moreover, T lymphocytes derived from a TREX1 heterozygous mutation showed an increased ability to inhibit neuroblastoma in vitro [106]. The tumor volume of TREX1 D18N mice was significantly smaller than that of WT mice, and the median survival time was also significantly prolonged [107], which proves that TREX1 inhibition on specific residues can be used as a viable target for anti-tumor therapy. Finding the best-targeted mutation sites that have the best inhibitory effect without causing severe autoimmune reactions may be the optimal choice for cancer immunotherapy. In other therapies activating the cGAS-STING pathway, STING agonists [108–112] have been used in anti-tumor immunity, which provides potential in TREX1 high-expression tumor therapy, although some drugs failed to pass clinical trials [113]. As an important second messenger, cGAMP not only activates the STING pathway in adjacent tumor cells through gap junctions [14] but can also be transported out of cells with the help of ABCC1. High expression of the ABCC1 transporter in tumor cells enhances anti-tumor immune responses by increasing cGAMP excretion into the tumor microenvironment [114]. However, ABCC1, also known as multidrug resistance protein, can also be resistant to some chemotherapy drugs, which are its export substrate [115]. Thus, drugs targeting TREX1 without the possibility of binding to ABCC1 may increase the anti-tumor effect.
Differently, TREX1 expression could be downregulated in some tumors. In the case of osteosarcoma, TREX1 expression is positively correlated with the prognosis—the lower the expression, the shorter the survival time. The low expression of the TREX1 gene may reduce the toxicity of CTL and NK cells to osteosarcoma cells, resulting in the immune escape of tumor cells [116]. It was found that non-stem cell osteosarcoma cells would acquire characteristics of tumor stem cells and promote tumor proliferation, differentiation, and metastasis after TREX1 gene knockdown. Inhibiting TREX1 may restrict the cell cycle and contribute to the production and renewal of tumor stem cells in tumor cells [117]. In addition, TREX1 expression was downregulated in human melanoma cell lines because TREX1 overexpression can induce apoptosis in melanoma cells and reduce tumor proliferation. It has been proven that carboplatin can activate TREX1 and reduce the proliferation of melanoma cancer cell lines, providing evidence for the treatment of malignant tumors with low TREX1 expression [118]. One possibility for the low TREX1 expression in tumor cells is that they have developed their own immune suppression system, inhibiting immune cells and suppressing the DNA damage response as well as TREX1 inhibition. The other possibility is that TREX1 deficiency results in the low TREX1 expression and immunosuppression. The AGS patients with TREX1-deficiency (especially the immunosuppressed ones) are more likely to have Hodgkin lymphoma or other hematological tumors [119], which confirms the possibility.
In a word, the discovery and application of TREX1 in tumor immunity merit further investigation to predict tumor growth, improve the efficacy of cytotoxic therapy combined with immunotherapy or targeted therapy, and contribute to the prognosis of patients with tumors.
Discussion
As a vital exonuclease in mammals, TREX1 not only degrades cytosolic DNA but also participates in glycosylation, ubiquitination, and DNA damage repair. In terms of protein composition, TREX1 exists as a homodimer, with different amino acid residues and domains playing dissimilar roles and cooperating with each other. Abnormalities in this key protein cause imbalances in DNA metabolic homeostasis, which leads to the accumulation of DNA debris in the cytosol, triggers the downstream inflammatory signal cascades and causes a variety of autoimmune diseases. The immunogenic self-DNA caused by TREX1 deficiency is primarily derived from cell death debris, genome replication waste, and retroelement activation. While the ssDNA produced by retroelement retrotransposition is sufficient to trigger an immune response, it only demonstrates that TREX1 and other enzymes are involved in the regulation of LINE-1. Current experiments are mostly limited to animal models and cell experiments, and the specific regulatory mechanisms or the differences between mice and humans are unknown, so expanded research is warranted. With the recent developments of gene-targeted therapy, medicines targeting TREX1 gene mutations may be able to treat related autoimmune diseases from the roots up. Exploring the sources of self-DNA contributes to targeted treatments for eliminating the antigens. However, due to the intricate genetic heterogeneity and various origins of antigens, treatments targeting these remain unsolved.
Thus, preventing the DNA-sensing downstream pathways, such as the cGAS-STING pathway, has provided a therapeutic target in recent years. Assorted studies have been carried out to investigate the treatments that target the cGAS-STING pathway. For now, inhibition of the cGAS mainly includes using small molecule drugs and nucleic acid drugs. A traditional small molecule, aspirin, could directly acetylate cGAS to inhibit its activity, which blunts cGAS-mediated interferon production in both AGS animal models and AGS patient cells [120]. In TREX1-deficient mice, a new antimalarial-like drug, X6, was shown to interact with the DNA-binding sites of cGAS and attenuate the cGAS-STING-mediated autoimmune responses [121]. More small molecule inhibitors of cGAS, such as RU.521 [122] and G compounds [123], are being identified through high-throughput screening assays, but whether they are effective in vivo remains unknown. In terms of nucleic acid-based drugs, microRNA (miR-23a/b) administration suppressed cGAS-mediated immune responses in TREX1−/− mice via their interaction with cGAS mRNA, though the treatment only mildly alleviated the animal model’s inflammatory phenotypes [124]. Anti-sense oligodeoxynucleotides A151 was discovered to effectively inhibit IFN production by blocking the way self-DNA binding with cGAS [125]. After being activated by DNA, cGAMP transfers to the STING and activates STING pathways. In the process, it was demonstrated that dorsomorphin could reduce cytosolic cGAMP accumulation and inhibit IFN production without affecting the STING-TBK1 pathway, which also effectively alleviates the disease phenotype of TREX1-deficient mice [126]. Because the downstream STING-TBK1-IRF3 pathway is a critical immune signal pathway in TREX1−/− or TREX1 mutant cells [127], it is a gold mine for immune targeted therapy. Canonical STING inhibitors mainly include the cyclic dinucleotide analogs, which replace or compete against the cGAMP with a higher affinity for the binding pocket of STING. For example, a cyclopeptide, astin C, specifically binds to STING, reduces the binding affinity of cGAMP to STING, suppresses IRF3 recruitment to STING, and attenuates autoinflammatory responses in TREX1−/− mice [128]. SN-011 binds to the STING with a higher affinity than the cGAMP and impedes STING oligomerization, trafficking and activation, inhibiting STING-mediated inflammation [129]. In addition, small molecules targeting specific residues of STING also show potential effects. C-178, C-176, and H-151 covalently target Cys-91 of STING and block the activation-induced palmitoylation of STING [130]. Palbociclib, a drug used to treat breast cancer, was found to directly bind to STING and disrupt its homodimerization by targeting Tyr-167, alleviating the autoimmune disease phenotype in TREX1−/− mice [131]. Because no STING-targeted drugs are currently approved for clinical use, repurposing the approved medicines may facilitate rapid development of clinical treatments for diseases induced by STING activation. Moreover, inhibiting the TBK1-mediated IFN response with Compound II [132], or targeting both IRF3 and NF-κB activation by Celastrol [133], provides more effective strategies for treating TREX1-related diseases. It is worth noting that the cGAS-STING pathway does not promote systemic autoimmunity in murine models of SLE, as shown in a previous study, but is rather dependent on endosomal TLRs [134]. Nonetheless, monogenic autoinflammatory diseases are believed to be driven by the aberrant accumulation of cytosolic DNA that is detected by the cGAS-STING pathway [13, 15, 19, 83]. Therefore, cGAS-STING-directed therapies may not be effective in systemic autoimmune diseases such as SLE, which provides important insights for the development of new therapies.
Because of the elevation of proinflammatory factors such as IFN-I in TREX1-deficient autoimmune diseases, the Janus kinases (JAKs) inflammatory signal cascade also becomes a potent target for treatment. A variety of JAKs inhibitors (jakinibs) have been reviewed [135], and despite the side effects, jakinibs could ameliorate the vicious inflammatory cascades in many diseases such as rheumatoid arthritis [136] and inflammatory bowel disease [137]. Inspiringly, jakinibs have been applied in the treatment of AGS [138], SLE [139], and FCL [140], which still requires more cohorts to evaluate the efficacy and safety of the therapy. A few studies of jakinib treatment have been reported in monogenic TREX1-deficient diseases. Specific inhibition (INCB039110) of Janus kinase 1 (JAK1, IFN-I receptor) has been shown to significantly reduce the expression of proinflammatory mediators, and topical treatment improves skin lesions in TREX1−/− mice [141]. A JAK1/2 inhibitor, baricitinib, was applied to treat three FCL patients with the TREX1 mutation. It significantly improved cutaneous lesions and partly relieved the pain of joint inflammation without severe adverse effects [142]. In addition, ruxolitinib [143] and tofacitinib [144] were respectively used in a single patient with TREX1-mutated FCL and AGS, showing positive results. Despite the small number of cases, successful treatments of jakinibs have also brought light to this spectrum of painful diseases that lack effective therapy. Taken together, targeting the inflammatory signal pathways of the self-DNA downstream might be a promising approach for TREX1-related autoimmune diseases (Fig. 5).
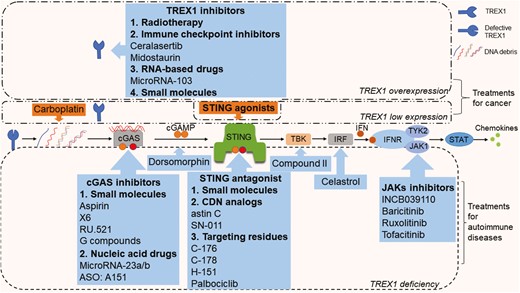
Potential treatments for TREX1-associated diseases. The blue callouts represent inhibition, and the orange callouts represent promotion. For tumors with TREX1 overexpression, therapies that inhibit TREX1 gene or protein expression and activate STING-related anti-tumor immune responses are recommended. For tumors with low TREX1 expression, treatment mainly activates the anti-tumor immunity. For autoimmune diseases, drugs targeting the cGAS-STING-TBK-IRF pathway and the JAK-STAT pathway have been found effective in experiments
In addition to autoimmune diseases, the role of TREX1 in cancer immunity also deserves more attention. As mentioned above, TREX1 overexpression reduces the immunogenicity of chromosomally unstable cells, prompting their transformation into cancer cells. The low TREX1 expression could be the consequence of immune suppression in some tumors. In turn, the tumor microenvironment may work to stimulate or inhibit TREX1 expression. TREX1 not only affects the growth and proliferation of tumor cells but can also be used as a target of immunotherapy and an indicator of the prognosis in patients with tumors. For tumor cells with TREX1 overexpression, silencing or reducing TREX1 gene expression indeed improves the immunogenicity of tumor cells, stimulates the anti-tumor immune response, and inhibits the growth of tumor cells. Some loss-of-function TREX1 mutant cells or animals also show resistance to tumor growth [106, 107]. Combining the aforementioned drugs, TREX1 inhibitory therapy mainly includes the following categories: radiotherapy [98], immunosuppressive drugs [100, 105], RNA-based drugs [104] and small molecules [107]. However, the use of drugs that inhibit TREX1 gene or protein expression will undoubtedly affect the function of TREX1 in normal cells and may increase the autoinflammatory response in animal models or humans. It is particularly important to find the key substances regulating TREX1 expression in tumor cells, which is different from that in normal cells. Although there is a lack of evidence for treating tumors with low TREX1 expression, activation of the downstream cGAS-STING pathway has also become a feasible therapy target for tumors with abnormal TREX1 expression. At present, targeted drugs mainly include small-molecule STING agonists [108–112]. Besides, the chemotherapeutic drug carboplatin has been shown to activate TREX1 and increase the expression of cGAS and STING [118]. It is possible that carboplatin leads to the modification of DNA structure and increases the accumulation of cytosolic DNA debris (Fig. 5). Due to the large number of tumor types and variable TREX1 expression in different kinds of cells, no uniform mechanism has yet been found for the role of TREX1 in cancer immunity. Future studies might pay more attention to the expression of TREX1 and related mechanisms in tumors, which will provide more possibilities in cancer immunotherapy.
In conclusion, this article focuses on the understanding of TREX1 structure, the source of self-DNA, and the mechanism of TREX1 degrading DNA. We summarized the TREX1-related autoimmune diseases and analyzed the connection between TREX1 abnormalities and cancer immunity. More in-depth studies on the TREX1 structure and gene sequence will be meaningful for discoveries in mechanisms and therapy targets.
Abbreviations
- AGS
Aicardi-Goutières syndrome
- ALR
AIM2-like receptor
- ATM
ataxia telangiectasia mutated
- cGAMP
cyclic GMP-AMP
- cGAS
cyclic GMP-AMP synthase
- CTL
cytotoxic T lymphocyte
- DAMP
damage-associated molecular pattern
- DCs
dendritic cells
- DEDD
Asp-Glu-Asp-Asp
- DNase III
deoxyribonuclease III
- dsDNA
double-stranded DNA
- ER
endoplasmic reticulum
- ERVs
endogenous retroviruses
- FCL
familial chilblain lupus
- HIV
human immunodeficiency virus
- IFI16
IFN-inducible 16
- IRF
interferon regulatory factor
- Jakinibs
JAKs inhibitors
- JAKs
Janus kinases
- LINE-1
long interspersed nuclear elements-1
- LLO
lipid-linked oligosaccharide
- LTR
long terminal repeat
- NETs
neutrophil extracellular traps
- NF-κB
nuclear factor kappa B
- NK
natural killer
- OST
oligosaccharide transferase
- PAMP
pathogen-associated molecular patterns
- PRR
pattern recognition receptor
- Rad51
recombination protein A
- RPA
replication protein A
- RVCL
retinal vasculopathy with cerebral leukodystrophy
- SAMHD1
SAM and HD domain-containing protein 1
- SLE
systemic lupus erythematosus
- ssDNA
single-stranded DNA
- STING
stimulator of interferon genes
- TLR
Toll-like receptor
- TREX1
three prime repair exonuclease 1.
Acknowledgements
We thank Kathleen Ding for checking grammar of the manuscript.
Ethical approval
Not applicable.
Conflict of interests
The authors declare that they have no competing interests.
Funding
This study was supported by the grants from the Scientific Research Foundation of the Institute for Translational Medicine of Anhui Province (No. 2017zhyx39), the Natural Science Foundation of Anhui Province (Nos. 1908085MH279 and 1508085MH158).
Author contributions
L.F., S.Y., and D.W. drafted the parts of TREX1 functions, DNA degradation, and autoimmune diseases sections. X.X. drafted the part about cancer immunity. L.F. and X.X. drafted all the figures. All authors contributed to writing, editing, and revising the manuscript.
Permission to reproduce (for relevant content)
Permission to copy, reproduce, or use the images should be obtained from the copyright owner and the authors based on reasonable requests.
Data availability
Not applicable.
References
Author notes
Fang, Ying and Xu. Contributed equally and share the first authorship.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}