




Abstract
The aim of this study was to clarify the significance of DNA methylation alterations during gastric carcinogenesis. Single-CpG resolution genome-wide DNA methylation analysis using the Infinium assay was performed on 109 samples of non-cancerous gastric mucosa (N) and 105 samples of tumorous tissue (T). DNA methylation alterations in T samples relative to N samples were evident for 3861 probes. Since N can be at the precancerous stage according to the field cancerization concept, unsupervised hierarchical clustering based on DNA methylation levels was performed on N samples (βN) using the 3861 probes. This divided the 109 patients into three clusters: A (n = 20), B1 (n = 20), and B2 (n = 69). Gastric carcinomas belonging to Cluster B1 showed tumor aggressiveness more frequently than those belonging to Clusters A and B2. The recurrence-free and overall survival rates of patients in Cluster B1 were lower than those of patients in Clusters A and B2. Sixty hallmark genes for which βN characterized the epigenetic clustering were identified. We then focused on DNA methylation levels in T samples (βT) of the 60 hallmark genes. In 48 of them, including the ADAM23, OLFM4, AMER2, GPSM1, CCL28, DTX1 and COL23A1 genes, βT was again significantly correlated with tumor aggressiveness, and the recurrence-free and/or overall survival rates. Multivariate analyses revealed that βT was a significant prognostic factor, being independent of clinicopathological parameters. These data indicate that DNA methylation profiles at the precancerous stage may be inherited by gastric carcinomas themselves, thus determining tumor aggressiveness and patient outcome.
Introduction
Gastric carcinoma is one of the most common malignancies worldwide (1). Despite improved surgical techniques and chemotherapy, patients with aggressive gastric carcinomas still have poor clinical outcomes (2). Therefore, there is a need to clarify the molecular backgrounds responsible for the clinicopathological diversity of gastric carcinomas. Oncogenic activation by mutations of the CTNNB1 (3) and PIK3CA (4) genes and amplification of the ERBB2 (5) gene, and inactivation of the CDH1 (6) and TP53 (7) tumor-suppressor genes by mutation, are frequent in gastric carcinomas. Recent whole-exome analysis has highlighted the significance of somatic mutation of the ARID1A gene in gastric carcinomas (8,9). However, such genetic alterations cannot fully explain the clinicopathological diversity of these malignancies.
As well as genetic alterations in gastric carcinomas, epigenetic changes have also been described (10,11); silencing of the CDH1 (12), CDKN2A (13), RUNX3 (14) and SFRP family (SFRP1, SFRP2 and SFRP5) genes (15) due to DNA hypermethylation around their promoter regions has been frequently observed. These tumor-suppressor genes are more frequently inactivated by aberrant DNA methylation than by genetic alterations, indicating the importance of DNA methylation during gastric carcinogenesis.
DNA methylation alterations are induced by carcinogenetic factors at the early and precancerous stage in various organs (16–18). With regard to the gastric mucosa, aberrant DNA methylation is reportedly induced by Helicobacter pylori (19) and Epstein–Barr (EB) virus infection (20), which are well-established factors associated with human gastric carcinogenesis. The concept of field cancerization in the stomach has now become established (21), which means that non-cancerous gastric mucosae obtained from patients with gastric carcinomas may be at the precancerous stage, following exposure to H.pylori, EB virus and other carcinogenetic factors. In organs other than the stomach, it has been suggested that DNA methylation profiles at the precancerous stage may determine tumor aggressiveness and patient outcome (16–18,22–26). However, it has still not been clarified whether correlations exist between DNA methylation profiles in non-cancerous gastric mucosae obtained from patients with gastric carcinomas and the clinicopathological aggressiveness of the carcinomas themselves, and subsequent outcome, in individual patients.
Although studies of gastric carcinomas (27,28) employing the single-CpG resolution Infinium array (29) have recently been published, they did not focus on DNA methylation in the non-cancerous mucosa. In this study, in order to clarify the significance of DNA methylation alterations at the precancerous stage of gastric carcinogenesis, we subjected 109 samples of non-cancerous mucosa (N) obtained from 109 patients with primary gastric carcinomas, and 105 samples of the corresponding tumorous tissues (T), to the Infinium assay.
Materials and methods
Patients and tissue samples
We employed 109 N samples and 105 T samples obtained from 109 patients with primary gastric carcinomas who underwent total or partial gastrectomy at the National Cancer Center Hospital, Japan. Tissue samples were immediately frozen and stored in liquid nitrogen until analysis. None of the patients had received any preoperative treatment. Among the patients, 79 were male and 30 were female, and their median age was 66 years (range, 26–91 years). Pathological staging and grading were based on the International Union Against Cancer classification (30). Histological types were determined based on the World Health Organization classification (31). All the tumors were classified according to the pathological tumor node metastasis (TNM) classification (32). Recurrence was diagnosed by clinicians on the basis of physical examination and imaging modalities such as computed tomography, magnetic resonance imaging, scintigraphy or positron emission tomography, and sometimes confirmed pathologically by biopsy. Clinicopathological parameters for the 109 patients are summarized in Supplementary Table 1 (available at Carcinogenesis Online).
Tissue specimens were provided by the National Cancer Center Biobank, Tokyo, Japan. This study was approved by the Ethics Committee of the National Cancer Center, Tokyo, Japan, and was performed in accordance with the Declaration of Helsinki. All patients included in this study provided written informed consent for the use of their materials.
Infinium assay
High-molecular weight DNA was extracted from fresh frozen tissue samples using phenol–chloroform, followed by dialysis. Five-hundred-nanogram aliquots of DNA were subjected to bisulfite conversion using an EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). DNA methylation status at 27578 CpG loci was examined at single-CpG resolution using the Infinium HumanMethylation27 Bead Array (Illumina, San Diego, CA). After hybridization, the specifically hybridized DNA was fluorescence-labeled by a single-base extension reaction and detected using a BeadScan reader (Illumina) in accordance with the manufacturer’s protocols. The data were then assembled using GenomeStudio methylation software (Illumina). At each CpG site, the ratio of the fluorescence signal was measured using a methylated probe relative to the sum of the methylated and unmethylated probes, i.e. the so-called β-value, which ranges from 0.00 to 1.00, reflecting the methylation level of an individual CpG site. The reliability of DNA methylation levels (β values) determined by Infinium assay has previously been verified using appropriate techniques such as pyrosequencing (QIAGEN GmbH, Hilden, Germany) (16,17,22).
Immunohistochemistry
Surgically resected materials of 107 patients, from whom formalin-fixed and paraffin-embedded tissue specimens were available, were subjected to immunohistochemistry. Five-micrometer-thick sections were deparaffinized, dehydrated and heated for 30min at 98°C in diluted Target retrieval solution, pH 9 (Dako, Carpinteria, CA) for antigen retrieval. Then all the sections were incubated with rabbit anti-H.pylori polyclonal antibody (Dako; dilution 1:50), and non-specific reactions were blocked with 2% normal swine serum. Primary antibody incubation was conducted at 4°C overnight, and was followed by incubation with EnVision+ Dual link system-HRP (Dako) at room temperature for 30min. 3.3′-Diaminobenzidine tetrahydrochloride was used as the chromogen. All sections were counterstained with hematoxylin. As a negative control, the primary antibody was omitted from the reaction sequence. Tissue specimens from patients in whom H.pylori infection had been detected by cultivation during clinical laboratory tests were used as positive controls.
Real-time quantitative RT-PCR analysis
Using TRIzol reagent (Life Technologies, Carlsbad, CA), total RNA was isolated from 33 N samples and 15 T samples, for which additional tissue specimens were available after DNA extraction. cDNA was reverse-transcribed from total RNA using random primers and Superscript III RNase H−Reverse Transcriptase (Life Technologies). Levels of expression of mRNA for the OLFM4, KCNQ5, FBN1, ITGA4 and ADAM23 genes were analyzed using custom TaqMan Expression Assays on the 7500 Fast Real-Time PCR System (Life Technologies) employing the relative standard curve method. The probes and PCR primer sets employed are summarized in Supplementary Table 2 (available at Carcinogenesis Online). Experiments were performed in triplicate, and the mean value for the three experiments was used as the CT value. All CT values were normalized to that of GAPDH in the same sample.
Statistics
In the Infinium assay, all CpG sites on chromosomes X and Y were excluded, to avoid any gender-specific methylation bias. In addition, the call proportions (P value of < 0.01 for detection of signals above the background) for 60 probes (shown in Supplementary Table 3, is available at Carcinogenesis Online) in the 109 N samples and 105 corresponding T samples (214 samples in total) were less than 90%. Since such a low proportion may be attributable to polymorphism at the probe CpG sites, these 60 probes were excluded from this assay, leaving a final total of 26426 autosomal CpG sites.
Infinium probes showing significant differences in DNA methylation levels between the 109 N samples and 105 T samples were identified by a logistic model adjusted by sex, age and experimental batch using Bonferroni correction (α = 3.78×10−7). Unsupervised hierarchical clustering (Euclidean distance, Ward’s method) based on DNA methylation levels in N samples (βN) was performed for the 109 patients. Correlations between clusters of patients and clinicopathological parameters were examined using Fisher’s exact test at a significance level of P < 0.05. Survival curves of patients belonging to each of the clusters obtained were generated by the Kaplan–Meier method, and the differences were compared by the log-rank test. The hallmark probes discriminating the clusters were identified by Welch’s t test using βN values.
Correlations between DNA methylation levels for the identified probes in T samples (βT) and the clinicopathological parameters of patients were examined using variance between groups (ANOVA) and Welch’s t test at a significance level of P < 0.05. The receiver operating characteristic (ROC) curve was generated and the Youden index of each probe was used as a cut-off value for examining correlations between DNA methylation levels and patient survival. Survival curves of patients belonging to groups showing higher (βT ≥ Youden index) and lower (βT < Youden index) DNA methylation levels were generated by the Kaplan–Meier method, and the differences were compared by the log-rank test. Multivariate analyses using the Cox proportional hazards regression model at a significance level of P < 0.05 were performed to examine the prognostic impact of clinicopathological parameters and DNA methylation levels (βT). All statistical analyses were performed using programming language R.
Results
Epigenetic clustering of gastric carcinomas based on DNA methylation profiles in N samples
In order to identify probes showing DNA methylation alterations associated with gastric carcinogenesis, we first employed the logistic model adjusted by sex, age and experimental batch for all 26426 probes. After Bonferroni correction (α = 3.78×10−7), 3861 probes (Supplementary Table 4, available at Carcinogenesis Online) showed significant differences in DNA methylation levels between the 109 N samples and 105 T samples.
Among the 3861 genes listed in Supplementary Table 4 (available at Carcinogenesis Online), DNA methylation data for 3404 obtained using more than 5 paired samples of N and T were deposited in the TCGA database (https://tcga-data.nci.nih.gov/tcga/). DNA hypermethylation (βT > βN) or hypomethylation (βT < βN) in T samples relative to N samples in our cohort of 3326 genes was found to be reproduced in the TCGA data, and such differences between N and T samples for 2145 genes reached statistically significant levels (P < 0.05), indicating that the DNA methylation profiles of gastric carcinomas in our cohort were generally validated by the TCGA data.
On the basis of the field cancerization concept, non-cancerous tissue obtained from patients with cancers derived from the same organs may be at the precancerous stage following exposure to carcinogenetic factors in vivo. In our previous studies of the kidney (22,23), lung (16,17), urinary bladder (24), liver (25) and pancreas (26), non-cancerous tissue from cancer patients frequently showed distinct DNA methylation profiles differing from those of normal tissue obtained from patients without cancer. Therefore, in this study, we focused on DNA methylation levels (βN) in N samples from the 109 patients with gastric carcinomas, and subjected them to unsupervised hierarchical clustering on the 3861 probes. This discriminated the patients into three clusters: A (n = 20), B1 (n = 20) and B2 (n = 69, Figure 1A).
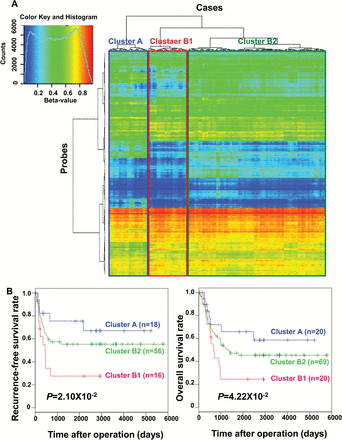
Epigenetic clustering of gastric carcinomas based on DNA methylation profiles in non-cancerous gastric mucosae (N). (A) Unsupervised hierarchical clustering (Euclidean distance, Ward’s method) using DNA methylation levels in N samples (βN) for the 3861 probes listed in Supplementary Table 4 (available at Carcinogenesis Online). Based on DNA methylation profiles in N samples (βN), all 109 patients with gastric cancers were subclustered into Cluster A (n = 20), Cluster B1 (n = 20) and Cluster B2 (n = 69). (B) Kaplan–Meier survival curves of patients belonging to Clusters A, B1 and B2. The period covered ranged from 4 to 5795 days (mean, 1611 days). In the 90 patients who underwent complete resection, the recurrence-free survival rate of patients in Cluster B1 was significantly lower than that of patients in Cluster A (P = 2.10×10-2, log-rank test). In all 109 patients, the overall survival rate of patients in Cluster B1 was significantly lower than that of patients in Cluster A (P = 4.22×10-2, log-rank test).
The clinicopathological parameters of the patients in these clusters based on βN are summarized in Table 1. Patients belonging to Cluster B1 were older than those belonging to Clusters A and B2, whereas the epigenetic clustering lacked any correlation with patient gender and H.pylori infection (Supplementary Figure 1, available at Carcinogenesis Online). The epigenetic clustering based on βN was significantly correlated with the clinicopathological parameters of the tumors: gastric carcinomas belonging to Cluster B1 more frequently showed undifferentiated histology, deeper invasion (higher pT stage) and a higher pathological TNM stage in comparison with gastric carcinomas belonging to Clusters A and B2 (Table 1). Gastric carcinomas belonging to Cluster B1 showed especially marked clinicopathological aggressiveness when compared to Cluster A.
Correlations between epigenetic clustering based on DNA methylation profiles in tissue specimens of non-cancerous gastric mucosa and clinicopathological parameters of the established gastric carcinomas
| Clinicopathological parameters | Cluster A | Cluster B1 | Cluster B2 | Pd |
|---|---|---|---|---|
| (n = 20) | (n = 20) | (n = 69) | ||
| Patients | ||||
| Age (years) | ||||
| ≥65 | 6 | 14 | 29 | 2.93×10−2 |
| <65 | 14 | 6 | 40 | |
| Sex | ||||
| Male | 17 | 15 | 47 | 3.45×10−1 |
| Female | 3 | 5 | 22 | |
| H.pylori infectiona | ||||
| Negative | 9 | 6 | 22 | 5.71×10−1 |
| Positive | 11 | 14 | 45 | |
| Gastric carcinomas | ||||
| Predominant histological classificationb | 3.07×10−2 | |||
| Differentiated | 15 | 7 | 34 | |
| Undifferentiated | 4 | 12 | 34 | |
| Mucin producing | 1 | 1 | 1 | |
| Most aggressive histological classificationc | 1.63×10−2 | |||
| Differentiated | 10 | 2 | 17 | |
| Undifferentiated | 10 | 18 | 52 | |
| Mucin-producing | 0 | 0 | 0 | |
| Tumor stage pT1–pT2 | 7 | 1 | 19 | 4.57×10−2 |
| pT3–pT4 | 13 | 19 | 50 | |
| Pathological tumor node metastasis stage | 8.78×10−3 | |||
| IA–IB | 7 | 0 | 18 | |
| IIA–IIB | 3 | 7 | 8 | |
| IIIA–IV | 10 | 13 | 43 | |
| Clinicopathological parameters | Cluster A | Cluster B1 | Cluster B2 | Pd |
|---|---|---|---|---|
| (n = 20) | (n = 20) | (n = 69) | ||
| Patients | ||||
| Age (years) | ||||
| ≥65 | 6 | 14 | 29 | 2.93×10−2 |
| <65 | 14 | 6 | 40 | |
| Sex | ||||
| Male | 17 | 15 | 47 | 3.45×10−1 |
| Female | 3 | 5 | 22 | |
| H.pylori infectiona | ||||
| Negative | 9 | 6 | 22 | 5.71×10−1 |
| Positive | 11 | 14 | 45 | |
| Gastric carcinomas | ||||
| Predominant histological classificationb | 3.07×10−2 | |||
| Differentiated | 15 | 7 | 34 | |
| Undifferentiated | 4 | 12 | 34 | |
| Mucin producing | 1 | 1 | 1 | |
| Most aggressive histological classificationc | 1.63×10−2 | |||
| Differentiated | 10 | 2 | 17 | |
| Undifferentiated | 10 | 18 | 52 | |
| Mucin-producing | 0 | 0 | 0 | |
| Tumor stage pT1–pT2 | 7 | 1 | 19 | 4.57×10−2 |
| pT3–pT4 | 13 | 19 | 50 | |
| Pathological tumor node metastasis stage | 8.78×10−3 | |||
| IA–IB | 7 | 0 | 18 | |
| IIA–IIB | 3 | 7 | 8 | |
| IIIA–IV | 10 | 13 | 43 | |
aImmunohistochemical examination was performed for 107 patients from whom formalin-fixed and paraffin-embedded tissue specimens were available.
bIf the tumor showed heterogeneity, findings in the predominant area were described.
cIf the tumor showed heterogeneity, the most aggressive features of the tumor were described.
dFisher’s exact test (P values of < 0.05 are underlined).
Correlations between epigenetic clustering based on DNA methylation profiles in tissue specimens of non-cancerous gastric mucosa and clinicopathological parameters of the established gastric carcinomas
| Clinicopathological parameters | Cluster A | Cluster B1 | Cluster B2 | Pd |
|---|---|---|---|---|
| (n = 20) | (n = 20) | (n = 69) | ||
| Patients | ||||
| Age (years) | ||||
| ≥65 | 6 | 14 | 29 | 2.93×10−2 |
| <65 | 14 | 6 | 40 | |
| Sex | ||||
| Male | 17 | 15 | 47 | 3.45×10−1 |
| Female | 3 | 5 | 22 | |
| H.pylori infectiona | ||||
| Negative | 9 | 6 | 22 | 5.71×10−1 |
| Positive | 11 | 14 | 45 | |
| Gastric carcinomas | ||||
| Predominant histological classificationb | 3.07×10−2 | |||
| Differentiated | 15 | 7 | 34 | |
| Undifferentiated | 4 | 12 | 34 | |
| Mucin producing | 1 | 1 | 1 | |
| Most aggressive histological classificationc | 1.63×10−2 | |||
| Differentiated | 10 | 2 | 17 | |
| Undifferentiated | 10 | 18 | 52 | |
| Mucin-producing | 0 | 0 | 0 | |
| Tumor stage pT1–pT2 | 7 | 1 | 19 | 4.57×10−2 |
| pT3–pT4 | 13 | 19 | 50 | |
| Pathological tumor node metastasis stage | 8.78×10−3 | |||
| IA–IB | 7 | 0 | 18 | |
| IIA–IIB | 3 | 7 | 8 | |
| IIIA–IV | 10 | 13 | 43 | |
| Clinicopathological parameters | Cluster A | Cluster B1 | Cluster B2 | Pd |
|---|---|---|---|---|
| (n = 20) | (n = 20) | (n = 69) | ||
| Patients | ||||
| Age (years) | ||||
| ≥65 | 6 | 14 | 29 | 2.93×10−2 |
| <65 | 14 | 6 | 40 | |
| Sex | ||||
| Male | 17 | 15 | 47 | 3.45×10−1 |
| Female | 3 | 5 | 22 | |
| H.pylori infectiona | ||||
| Negative | 9 | 6 | 22 | 5.71×10−1 |
| Positive | 11 | 14 | 45 | |
| Gastric carcinomas | ||||
| Predominant histological classificationb | 3.07×10−2 | |||
| Differentiated | 15 | 7 | 34 | |
| Undifferentiated | 4 | 12 | 34 | |
| Mucin producing | 1 | 1 | 1 | |
| Most aggressive histological classificationc | 1.63×10−2 | |||
| Differentiated | 10 | 2 | 17 | |
| Undifferentiated | 10 | 18 | 52 | |
| Mucin-producing | 0 | 0 | 0 | |
| Tumor stage pT1–pT2 | 7 | 1 | 19 | 4.57×10−2 |
| pT3–pT4 | 13 | 19 | 50 | |
| Pathological tumor node metastasis stage | 8.78×10−3 | |||
| IA–IB | 7 | 0 | 18 | |
| IIA–IIB | 3 | 7 | 8 | |
| IIIA–IV | 10 | 13 | 43 | |
aImmunohistochemical examination was performed for 107 patients from whom formalin-fixed and paraffin-embedded tissue specimens were available.
bIf the tumor showed heterogeneity, findings in the predominant area were described.
cIf the tumor showed heterogeneity, the most aggressive features of the tumor were described.
dFisher’s exact test (P values of < 0.05 are underlined).
Figure 1B shows the Kaplan–Meier survival curves of patients belonging to Clusters A, B1 and B2. The period covered ranged from 4 to 5795 days (mean, 1611 days). In the 90 patients who underwent complete resection, the recurrence-free survival rate for Cluster B1 was significantly lower than that for Cluster A (P = 2.10×10−2). The overall survival rate for Cluster B1 patients was significantly lower than that for Cluster A patients (P = 4.22×10−2, log-rank test).
In order to identify those probes whose DNA methylation status characterized the epigenetic clustering based on βN, i.e. those showing significant differences between the most aggressive Cluster B1 and the least-aggressive Cluster A, Welch’s t test was performed. This revealed that 3249 and 6418 probes showed significantly higher and lower DNA methylation levels in N samples (βN) of Cluster B1 than βN of Cluster A, respectively (P < 0.05, Welch’s t test). Among 3249 probes that showed significantly higher βN values in Cluster B1 than in Cluster A, the top 30 showing the largest differences in βN values between the two clusters are listed in Table 2A (Supplementary Figure 2, available at Carcinogenesis Online). Among 6418 probes that showed significantly lower βN values in Cluster B1 than in Cluster A, the top 30 showing the largest differences in βN values between the two clusters are listed in Table 2B (Supplementary Figure 2, available at Carcinogenesis Online).
Top 60 probes showing DNA methylation status characterizing the epigenetic clustering
(A) Top 30 probes showing significant DNA hypermethylation in N samples of Cluster B1 compared to those of Cluster A (P < 0.05, Welch’s t test) and the largest differences in average DNA methylation levels between Clusters B1 and A (ΔβB1-A)
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean±SD) | P | ΔβB1–A | |
|---|---|---|---|---|---|---|---|
| Cluster A | ClusterB1 | ||||||
| cg23743114 | 17 | 34328396 | CCL15-CCL14 | 0.385±0.091 | 0.689±0.055 | 1.31×10−16 | 0.304 |
| cg02192965 | 2 | 44502740 | SLC3A1 | 0.417±0.091 | 0.708±0.048 | 6.47×10−19 | 0.291 |
| cg18754342 | 12 | 14849268 | GUCY2C | 0.444±0.088 | 0.732±0.046 | 2.71×10−14 | 0.288 |
| cg14934821 | 9 | 139228820 | GPSM1 | 0.480±0.068 | 0.758±0.052 | 2.37×10−17 | 0.279 |
| cg07220939 | 11 | 64358617 | SLC22A12 | 0.419±0.118 | 0.697±0.051 | 1.41×10−16 | 0.279 |
| cg26530341 | 8 | 23083353 | LOC389641 | 0.409±0.067 | 0.686±0.072 | 8.32×10−19 | 0.277 |
| cg04968426 | 15 | 41120711 | PPP1R14D | 0.581±0.085 | 0.856±0.041 | 1.89×10−20 | 0.276 |
| cg03364504 | 1 | 113393176 | LOC100996702 | 0.598±0.077 | 0.871±0.034 | 3.14×10−14 | 0.273 |
| cg03545635 | 7 | 2471551 | CHST12 | 0.514±0.102 | 0.784±0.041 | 6.72×10−15 | 0.270 |
| cg07150830 | 17 | 26127542 | NOS2 | 0.627±0.087 | 0.897±0.039 | 1.23×10−13 | 0.270 |
| cg12038710 | 8 | 95220583 | CDH17 | 0.577±0.085 | 0.842±0.030 | 5.48×10−11 | 0.265 |
| cg21375825 | 2 | 136594646 | LCT | 0.559±0.119 | 0.825±0.028 | 3.84×10−14 | 0.265 |
| cg12582008 | 13 | 53603286 | OLFM4 | 0.542±0.079 | 0.807±0.066 | 9.84×10−15 | 0.264 |
| cg03016571 | 17 | 48844124 | LINC00483 | 0.463±0.087 | 0.722±0.054 | 1.45×10−15 | 0.259 |
| cg21122774 | 9 | 136604996 | SARDH | 0.514±0.091 | 0.770±0.038 | 5.20×10−10 | 0.255 |
| cg17778120 | 3 | 139195319 | RBP2 | 0.557±0.086 | 0.811±0.034 | 1.41×10−10 | 0.254 |
| cg09081544 | 3 | 124652790 | MUC13 | 0.483±0.074 | 0.736±0.048 | 2.19×10−11 | 0.252 |
| cg09448875 | 10 | 101542449 | ABCC2 | 0.480±0.083 | 0.727±0.061 | 3.92×10−16 | 0.247 |
| cg03077492 | 5 | 43413095 | CCL28 | 0.602±0.093 | 0.849±0.038 | 8.84×10−13 | 0.247 |
| cg24027679 | 1 | 9086621 | SLC2A7 | 0.579±0.103 | 0.825±0.045 | 2.58×10-10 | 0.246 |
| cg18971054 | 7 | 141695759 | MGAM | 0.552±0.090 | 0.795±0.032 | 6.16×10−13 | 0.243 |
| cg03483654 | 11 | 61102074 | DDB1 | 0.719±0.108 | 0.962±0.025 | 8.20×10−16 | 0.243 |
| cg02044879 | 10 | 74714935 | PLA2G12B | 0.510±0.076 | 0.752±0.038 | 7.45×10−18 | 0.243 |
| cg20683151 | 2 | 228243v972 | TM4SF20 | 0.606±0.090 | 0.848±0.025 | 1.10×10−18 | 0.242 |
| cg06665322 | 1 | 167059365 | GPA33 | 0.452±0.088 | 0.693±0.051 | 7.03×10−17 | 0.242 |
| cg17142183 | 2 | 102608192 | IL1R2 | 0.501±0.084 | 0.742±0.053 | 2.57×10−16 | 0.241 |
| cg21591452 | 17 | 79304628 | TMEM105 | 0.669±0.070 | 0.908±0.032 | 1.57×10−11 | 0.239 |
| cg06277277 | 1 | 161208307 | NR1I3 | 0.519±0.099 | 0.751±0.035 | 5.22×10−15 | 0.231 |
| cg11920519 | 20 | 33135025 | MAP1LC3A | 0.587±0.072 | 0.817±0.049 | 2.47×10−14 | 0.229 |
| cg16575408 | 11 | 102669291 | MMP1 | 0.672±0.099 | 0.899±0.029 | 3.75×10−9 | 0.228 |
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean±SD) | P | ΔβB1–A | |
|---|---|---|---|---|---|---|---|
| Cluster A | ClusterB1 | ||||||
| cg23743114 | 17 | 34328396 | CCL15-CCL14 | 0.385±0.091 | 0.689±0.055 | 1.31×10−16 | 0.304 |
| cg02192965 | 2 | 44502740 | SLC3A1 | 0.417±0.091 | 0.708±0.048 | 6.47×10−19 | 0.291 |
| cg18754342 | 12 | 14849268 | GUCY2C | 0.444±0.088 | 0.732±0.046 | 2.71×10−14 | 0.288 |
| cg14934821 | 9 | 139228820 | GPSM1 | 0.480±0.068 | 0.758±0.052 | 2.37×10−17 | 0.279 |
| cg07220939 | 11 | 64358617 | SLC22A12 | 0.419±0.118 | 0.697±0.051 | 1.41×10−16 | 0.279 |
| cg26530341 | 8 | 23083353 | LOC389641 | 0.409±0.067 | 0.686±0.072 | 8.32×10−19 | 0.277 |
| cg04968426 | 15 | 41120711 | PPP1R14D | 0.581±0.085 | 0.856±0.041 | 1.89×10−20 | 0.276 |
| cg03364504 | 1 | 113393176 | LOC100996702 | 0.598±0.077 | 0.871±0.034 | 3.14×10−14 | 0.273 |
| cg03545635 | 7 | 2471551 | CHST12 | 0.514±0.102 | 0.784±0.041 | 6.72×10−15 | 0.270 |
| cg07150830 | 17 | 26127542 | NOS2 | 0.627±0.087 | 0.897±0.039 | 1.23×10−13 | 0.270 |
| cg12038710 | 8 | 95220583 | CDH17 | 0.577±0.085 | 0.842±0.030 | 5.48×10−11 | 0.265 |
| cg21375825 | 2 | 136594646 | LCT | 0.559±0.119 | 0.825±0.028 | 3.84×10−14 | 0.265 |
| cg12582008 | 13 | 53603286 | OLFM4 | 0.542±0.079 | 0.807±0.066 | 9.84×10−15 | 0.264 |
| cg03016571 | 17 | 48844124 | LINC00483 | 0.463±0.087 | 0.722±0.054 | 1.45×10−15 | 0.259 |
| cg21122774 | 9 | 136604996 | SARDH | 0.514±0.091 | 0.770±0.038 | 5.20×10−10 | 0.255 |
| cg17778120 | 3 | 139195319 | RBP2 | 0.557±0.086 | 0.811±0.034 | 1.41×10−10 | 0.254 |
| cg09081544 | 3 | 124652790 | MUC13 | 0.483±0.074 | 0.736±0.048 | 2.19×10−11 | 0.252 |
| cg09448875 | 10 | 101542449 | ABCC2 | 0.480±0.083 | 0.727±0.061 | 3.92×10−16 | 0.247 |
| cg03077492 | 5 | 43413095 | CCL28 | 0.602±0.093 | 0.849±0.038 | 8.84×10−13 | 0.247 |
| cg24027679 | 1 | 9086621 | SLC2A7 | 0.579±0.103 | 0.825±0.045 | 2.58×10-10 | 0.246 |
| cg18971054 | 7 | 141695759 | MGAM | 0.552±0.090 | 0.795±0.032 | 6.16×10−13 | 0.243 |
| cg03483654 | 11 | 61102074 | DDB1 | 0.719±0.108 | 0.962±0.025 | 8.20×10−16 | 0.243 |
| cg02044879 | 10 | 74714935 | PLA2G12B | 0.510±0.076 | 0.752±0.038 | 7.45×10−18 | 0.243 |
| cg20683151 | 2 | 228243v972 | TM4SF20 | 0.606±0.090 | 0.848±0.025 | 1.10×10−18 | 0.242 |
| cg06665322 | 1 | 167059365 | GPA33 | 0.452±0.088 | 0.693±0.051 | 7.03×10−17 | 0.242 |
| cg17142183 | 2 | 102608192 | IL1R2 | 0.501±0.084 | 0.742±0.053 | 2.57×10−16 | 0.241 |
| cg21591452 | 17 | 79304628 | TMEM105 | 0.669±0.070 | 0.908±0.032 | 1.57×10−11 | 0.239 |
| cg06277277 | 1 | 161208307 | NR1I3 | 0.519±0.099 | 0.751±0.035 | 5.22×10−15 | 0.231 |
| cg11920519 | 20 | 33135025 | MAP1LC3A | 0.587±0.072 | 0.817±0.049 | 2.47×10−14 | 0.229 |
| cg16575408 | 11 | 102669291 | MMP1 | 0.672±0.099 | 0.899±0.029 | 3.75×10−9 | 0.228 |
Top 60 probes showing DNA methylation status characterizing the epigenetic clustering
(A) Top 30 probes showing significant DNA hypermethylation in N samples of Cluster B1 compared to those of Cluster A (P < 0.05, Welch’s t test) and the largest differences in average DNA methylation levels between Clusters B1 and A (ΔβB1-A)
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean±SD) | P | ΔβB1–A | |
|---|---|---|---|---|---|---|---|
| Cluster A | ClusterB1 | ||||||
| cg23743114 | 17 | 34328396 | CCL15-CCL14 | 0.385±0.091 | 0.689±0.055 | 1.31×10−16 | 0.304 |
| cg02192965 | 2 | 44502740 | SLC3A1 | 0.417±0.091 | 0.708±0.048 | 6.47×10−19 | 0.291 |
| cg18754342 | 12 | 14849268 | GUCY2C | 0.444±0.088 | 0.732±0.046 | 2.71×10−14 | 0.288 |
| cg14934821 | 9 | 139228820 | GPSM1 | 0.480±0.068 | 0.758±0.052 | 2.37×10−17 | 0.279 |
| cg07220939 | 11 | 64358617 | SLC22A12 | 0.419±0.118 | 0.697±0.051 | 1.41×10−16 | 0.279 |
| cg26530341 | 8 | 23083353 | LOC389641 | 0.409±0.067 | 0.686±0.072 | 8.32×10−19 | 0.277 |
| cg04968426 | 15 | 41120711 | PPP1R14D | 0.581±0.085 | 0.856±0.041 | 1.89×10−20 | 0.276 |
| cg03364504 | 1 | 113393176 | LOC100996702 | 0.598±0.077 | 0.871±0.034 | 3.14×10−14 | 0.273 |
| cg03545635 | 7 | 2471551 | CHST12 | 0.514±0.102 | 0.784±0.041 | 6.72×10−15 | 0.270 |
| cg07150830 | 17 | 26127542 | NOS2 | 0.627±0.087 | 0.897±0.039 | 1.23×10−13 | 0.270 |
| cg12038710 | 8 | 95220583 | CDH17 | 0.577±0.085 | 0.842±0.030 | 5.48×10−11 | 0.265 |
| cg21375825 | 2 | 136594646 | LCT | 0.559±0.119 | 0.825±0.028 | 3.84×10−14 | 0.265 |
| cg12582008 | 13 | 53603286 | OLFM4 | 0.542±0.079 | 0.807±0.066 | 9.84×10−15 | 0.264 |
| cg03016571 | 17 | 48844124 | LINC00483 | 0.463±0.087 | 0.722±0.054 | 1.45×10−15 | 0.259 |
| cg21122774 | 9 | 136604996 | SARDH | 0.514±0.091 | 0.770±0.038 | 5.20×10−10 | 0.255 |
| cg17778120 | 3 | 139195319 | RBP2 | 0.557±0.086 | 0.811±0.034 | 1.41×10−10 | 0.254 |
| cg09081544 | 3 | 124652790 | MUC13 | 0.483±0.074 | 0.736±0.048 | 2.19×10−11 | 0.252 |
| cg09448875 | 10 | 101542449 | ABCC2 | 0.480±0.083 | 0.727±0.061 | 3.92×10−16 | 0.247 |
| cg03077492 | 5 | 43413095 | CCL28 | 0.602±0.093 | 0.849±0.038 | 8.84×10−13 | 0.247 |
| cg24027679 | 1 | 9086621 | SLC2A7 | 0.579±0.103 | 0.825±0.045 | 2.58×10-10 | 0.246 |
| cg18971054 | 7 | 141695759 | MGAM | 0.552±0.090 | 0.795±0.032 | 6.16×10−13 | 0.243 |
| cg03483654 | 11 | 61102074 | DDB1 | 0.719±0.108 | 0.962±0.025 | 8.20×10−16 | 0.243 |
| cg02044879 | 10 | 74714935 | PLA2G12B | 0.510±0.076 | 0.752±0.038 | 7.45×10−18 | 0.243 |
| cg20683151 | 2 | 228243v972 | TM4SF20 | 0.606±0.090 | 0.848±0.025 | 1.10×10−18 | 0.242 |
| cg06665322 | 1 | 167059365 | GPA33 | 0.452±0.088 | 0.693±0.051 | 7.03×10−17 | 0.242 |
| cg17142183 | 2 | 102608192 | IL1R2 | 0.501±0.084 | 0.742±0.053 | 2.57×10−16 | 0.241 |
| cg21591452 | 17 | 79304628 | TMEM105 | 0.669±0.070 | 0.908±0.032 | 1.57×10−11 | 0.239 |
| cg06277277 | 1 | 161208307 | NR1I3 | 0.519±0.099 | 0.751±0.035 | 5.22×10−15 | 0.231 |
| cg11920519 | 20 | 33135025 | MAP1LC3A | 0.587±0.072 | 0.817±0.049 | 2.47×10−14 | 0.229 |
| cg16575408 | 11 | 102669291 | MMP1 | 0.672±0.099 | 0.899±0.029 | 3.75×10−9 | 0.228 |
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean±SD) | P | ΔβB1–A | |
|---|---|---|---|---|---|---|---|
| Cluster A | ClusterB1 | ||||||
| cg23743114 | 17 | 34328396 | CCL15-CCL14 | 0.385±0.091 | 0.689±0.055 | 1.31×10−16 | 0.304 |
| cg02192965 | 2 | 44502740 | SLC3A1 | 0.417±0.091 | 0.708±0.048 | 6.47×10−19 | 0.291 |
| cg18754342 | 12 | 14849268 | GUCY2C | 0.444±0.088 | 0.732±0.046 | 2.71×10−14 | 0.288 |
| cg14934821 | 9 | 139228820 | GPSM1 | 0.480±0.068 | 0.758±0.052 | 2.37×10−17 | 0.279 |
| cg07220939 | 11 | 64358617 | SLC22A12 | 0.419±0.118 | 0.697±0.051 | 1.41×10−16 | 0.279 |
| cg26530341 | 8 | 23083353 | LOC389641 | 0.409±0.067 | 0.686±0.072 | 8.32×10−19 | 0.277 |
| cg04968426 | 15 | 41120711 | PPP1R14D | 0.581±0.085 | 0.856±0.041 | 1.89×10−20 | 0.276 |
| cg03364504 | 1 | 113393176 | LOC100996702 | 0.598±0.077 | 0.871±0.034 | 3.14×10−14 | 0.273 |
| cg03545635 | 7 | 2471551 | CHST12 | 0.514±0.102 | 0.784±0.041 | 6.72×10−15 | 0.270 |
| cg07150830 | 17 | 26127542 | NOS2 | 0.627±0.087 | 0.897±0.039 | 1.23×10−13 | 0.270 |
| cg12038710 | 8 | 95220583 | CDH17 | 0.577±0.085 | 0.842±0.030 | 5.48×10−11 | 0.265 |
| cg21375825 | 2 | 136594646 | LCT | 0.559±0.119 | 0.825±0.028 | 3.84×10−14 | 0.265 |
| cg12582008 | 13 | 53603286 | OLFM4 | 0.542±0.079 | 0.807±0.066 | 9.84×10−15 | 0.264 |
| cg03016571 | 17 | 48844124 | LINC00483 | 0.463±0.087 | 0.722±0.054 | 1.45×10−15 | 0.259 |
| cg21122774 | 9 | 136604996 | SARDH | 0.514±0.091 | 0.770±0.038 | 5.20×10−10 | 0.255 |
| cg17778120 | 3 | 139195319 | RBP2 | 0.557±0.086 | 0.811±0.034 | 1.41×10−10 | 0.254 |
| cg09081544 | 3 | 124652790 | MUC13 | 0.483±0.074 | 0.736±0.048 | 2.19×10−11 | 0.252 |
| cg09448875 | 10 | 101542449 | ABCC2 | 0.480±0.083 | 0.727±0.061 | 3.92×10−16 | 0.247 |
| cg03077492 | 5 | 43413095 | CCL28 | 0.602±0.093 | 0.849±0.038 | 8.84×10−13 | 0.247 |
| cg24027679 | 1 | 9086621 | SLC2A7 | 0.579±0.103 | 0.825±0.045 | 2.58×10-10 | 0.246 |
| cg18971054 | 7 | 141695759 | MGAM | 0.552±0.090 | 0.795±0.032 | 6.16×10−13 | 0.243 |
| cg03483654 | 11 | 61102074 | DDB1 | 0.719±0.108 | 0.962±0.025 | 8.20×10−16 | 0.243 |
| cg02044879 | 10 | 74714935 | PLA2G12B | 0.510±0.076 | 0.752±0.038 | 7.45×10−18 | 0.243 |
| cg20683151 | 2 | 228243v972 | TM4SF20 | 0.606±0.090 | 0.848±0.025 | 1.10×10−18 | 0.242 |
| cg06665322 | 1 | 167059365 | GPA33 | 0.452±0.088 | 0.693±0.051 | 7.03×10−17 | 0.242 |
| cg17142183 | 2 | 102608192 | IL1R2 | 0.501±0.084 | 0.742±0.053 | 2.57×10−16 | 0.241 |
| cg21591452 | 17 | 79304628 | TMEM105 | 0.669±0.070 | 0.908±0.032 | 1.57×10−11 | 0.239 |
| cg06277277 | 1 | 161208307 | NR1I3 | 0.519±0.099 | 0.751±0.035 | 5.22×10−15 | 0.231 |
| cg11920519 | 20 | 33135025 | MAP1LC3A | 0.587±0.072 | 0.817±0.049 | 2.47×10−14 | 0.229 |
| cg16575408 | 11 | 102669291 | MMP1 | 0.672±0.099 | 0.899±0.029 | 3.75×10−9 | 0.228 |
(B) Top 30 probes showing significant DNA hypomethylation in N samples of Cluster B1 compared to those of Cluster A (P < 0.05, Welch’s t test) and the largest differences in average DNA methylation levels between Clusters B1 and A (ΔβA–B1).
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean ± SD) | P | ΔβA–B1 | |
|---|---|---|---|---|---|---|---|
| Cluster A | Cluster B1 | ||||||
| cg24687051 | 6 | 73332073 | KCNQ5 | 0.543±0.108 | 0.059±0.051 | 1.31×10−16 | 0.484 |
| cg03168582 | 9 | 841850 | DMRT1 | 0.588±0.074 | 0.155±0.088 | 6.47×10−19 | 0.433 |
| cg17892556 | 19 | 12267464 | ZNF625 | 0.523±0.112 | 0.120±0.051 | 2.71×10−14 | 0.403 |
| cg07080358 | 2 | 68546507 | CNRIP1 | 0.542±0.092 | 0.142±0.067 | 2.37×10−17 | 0.400 |
| cg26309134 | 19 | 56879571 | ZNF542 | 0.519±0.092 | 0.121±0.048 | 1.41×10−16 | 0.398 |
| cg11657808 | 1 | 237205950 | RYR2 | 0.626±0.080 | 0.238±0.066 | 8.32×10−19 | 0.388 |
| cg11939071 | 12 | 113494429 | DTX1 | 0.465±0.070 | 0.084±0.053 | 1.89×10−20 | 0.381 |
| cg22029275 | 13 | 25745784 | AMER2 | 0.522±0.109 | 0.141±0.057 | 3.14×10−14 | 0.380 |
| cg22619018 | 8 | 4852624 | CSMD1 | 0.738±0.077 | 0.357±0.106 | 6.72×10−15 | 0.380 |
| cg12629325 | 5 | 140306458 | PCDHAC1 | 0.705±0.062 | 0.325±0.115 | 1.23×10−13 | 0.380 |
| cg17872757 | 11 | 128564180 | FLI1 | 0.439±0.135 | 0.066±0.034 | 5.48×10−11 | 0.373 |
| cg07017374 | 13 | 28674451 | FLT3 | 0.519±0.107 | 0.154±0.084 | 3.84×10−14 | 0.365 |
| cg13562911 | 6 | 11044106 | ELOVL2-AS1 | 0.501±0.101 | 0.139±0.060 | 9.84×10−15 | 0.362 |
| cg18671950 | 15 | 48936953 | FBN1 | 0.522±0.063 | 0.166±0.094 | 1.45×10−15 | 0.356 |
| cg06744574 | 1 | 49242359 | BEND5 | 0.417±0.150 | 0.065±0.064 | 5.20×10−10 | 0.352 |
| cg19118812 | 7 | 37488438 | ELMO1 | 0.417±0.137 | 0.066±0.046 | 1.41×10−10 | 0.350 |
| cg12874092 | 10 | 17271519 | VIM | 0.379±0.115 | 0.030±0.015 | 2.19×10−11 | 0.349 |
| cg09551147 | 10 | 106399957 | SORCS3 | 0.479±0.086 | 0.138±0.058 | 3.92×10−16 | 0.341 |
| cg25583174 | 4 | 123748386 | FGF2 | 0.439±0.112 | 0.099±0.064 | 8.84×10−13 | 0.340 |
| cg04034767 | 12 | 52400907 | GRASP | 0.453±0.136 | 0.114±0.046 | 2.58×10−10 | 0.338 |
| cg17525406 | 1 | 4715520 | AJAP1 | 0.709±0.068 | 0.372±0.111 | 6.16×10−13 | 0.337 |
| cg25886284 | 19 | 36909418 | ZFP82 | 0.459±0.084 | 0.122±0.075 | 8.20×10−16 | 0.336 |
| cg21475402 | 1 | 156612140 | BCAN | 0.569±0.074 | 0.232±0.059 | 7.45×10−18 | 0.336 |
| cg08383315 | 11 | 8190565 | RIC3 | 0.475±0.070 | 0.139±0.054 | 1.10×10−18 | 0.336 |
| cg21790626 | 19 | 58220494 | ZNF154 | 0.552±0.066 | 0.217±0.079 | 7.03×10−17 | 0.335 |
| cg16787600 | 10 | 106400880 | SORCS3 | 0.623±0.072 | 0.288±0.081 | 2.57×10−16 | 0.335 |
| cg20415809 | 2 | 182321855 | ITGA4 | 0.434±0.118 | 0.102±0.044 | 1.57×10−11 | 0.332 |
| cg10730712 | 5 | 178017827 | COL23A1 | 0.497±0.091 | 0.166±0.055 | 5.22×10−15 | 0.331 |
| cg01775414 | 22 | 45405404 | PHF21B | 0.496±0.096 | 0.165±0.062 | 2.47×10−14 | 0.330 |
| cg16778809 | 2 | 207308375 | ADAM23 | 0.438±0.155 | 0.108±0.061 | 3.75×10−09 | 0.330 |
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean ± SD) | P | ΔβA–B1 | |
|---|---|---|---|---|---|---|---|
| Cluster A | Cluster B1 | ||||||
| cg24687051 | 6 | 73332073 | KCNQ5 | 0.543±0.108 | 0.059±0.051 | 1.31×10−16 | 0.484 |
| cg03168582 | 9 | 841850 | DMRT1 | 0.588±0.074 | 0.155±0.088 | 6.47×10−19 | 0.433 |
| cg17892556 | 19 | 12267464 | ZNF625 | 0.523±0.112 | 0.120±0.051 | 2.71×10−14 | 0.403 |
| cg07080358 | 2 | 68546507 | CNRIP1 | 0.542±0.092 | 0.142±0.067 | 2.37×10−17 | 0.400 |
| cg26309134 | 19 | 56879571 | ZNF542 | 0.519±0.092 | 0.121±0.048 | 1.41×10−16 | 0.398 |
| cg11657808 | 1 | 237205950 | RYR2 | 0.626±0.080 | 0.238±0.066 | 8.32×10−19 | 0.388 |
| cg11939071 | 12 | 113494429 | DTX1 | 0.465±0.070 | 0.084±0.053 | 1.89×10−20 | 0.381 |
| cg22029275 | 13 | 25745784 | AMER2 | 0.522±0.109 | 0.141±0.057 | 3.14×10−14 | 0.380 |
| cg22619018 | 8 | 4852624 | CSMD1 | 0.738±0.077 | 0.357±0.106 | 6.72×10−15 | 0.380 |
| cg12629325 | 5 | 140306458 | PCDHAC1 | 0.705±0.062 | 0.325±0.115 | 1.23×10−13 | 0.380 |
| cg17872757 | 11 | 128564180 | FLI1 | 0.439±0.135 | 0.066±0.034 | 5.48×10−11 | 0.373 |
| cg07017374 | 13 | 28674451 | FLT3 | 0.519±0.107 | 0.154±0.084 | 3.84×10−14 | 0.365 |
| cg13562911 | 6 | 11044106 | ELOVL2-AS1 | 0.501±0.101 | 0.139±0.060 | 9.84×10−15 | 0.362 |
| cg18671950 | 15 | 48936953 | FBN1 | 0.522±0.063 | 0.166±0.094 | 1.45×10−15 | 0.356 |
| cg06744574 | 1 | 49242359 | BEND5 | 0.417±0.150 | 0.065±0.064 | 5.20×10−10 | 0.352 |
| cg19118812 | 7 | 37488438 | ELMO1 | 0.417±0.137 | 0.066±0.046 | 1.41×10−10 | 0.350 |
| cg12874092 | 10 | 17271519 | VIM | 0.379±0.115 | 0.030±0.015 | 2.19×10−11 | 0.349 |
| cg09551147 | 10 | 106399957 | SORCS3 | 0.479±0.086 | 0.138±0.058 | 3.92×10−16 | 0.341 |
| cg25583174 | 4 | 123748386 | FGF2 | 0.439±0.112 | 0.099±0.064 | 8.84×10−13 | 0.340 |
| cg04034767 | 12 | 52400907 | GRASP | 0.453±0.136 | 0.114±0.046 | 2.58×10−10 | 0.338 |
| cg17525406 | 1 | 4715520 | AJAP1 | 0.709±0.068 | 0.372±0.111 | 6.16×10−13 | 0.337 |
| cg25886284 | 19 | 36909418 | ZFP82 | 0.459±0.084 | 0.122±0.075 | 8.20×10−16 | 0.336 |
| cg21475402 | 1 | 156612140 | BCAN | 0.569±0.074 | 0.232±0.059 | 7.45×10−18 | 0.336 |
| cg08383315 | 11 | 8190565 | RIC3 | 0.475±0.070 | 0.139±0.054 | 1.10×10−18 | 0.336 |
| cg21790626 | 19 | 58220494 | ZNF154 | 0.552±0.066 | 0.217±0.079 | 7.03×10−17 | 0.335 |
| cg16787600 | 10 | 106400880 | SORCS3 | 0.623±0.072 | 0.288±0.081 | 2.57×10−16 | 0.335 |
| cg20415809 | 2 | 182321855 | ITGA4 | 0.434±0.118 | 0.102±0.044 | 1.57×10−11 | 0.332 |
| cg10730712 | 5 | 178017827 | COL23A1 | 0.497±0.091 | 0.166±0.055 | 5.22×10−15 | 0.331 |
| cg01775414 | 22 | 45405404 | PHF21B | 0.496±0.096 | 0.165±0.062 | 2.47×10−14 | 0.330 |
| cg16778809 | 2 | 207308375 | ADAM23 | 0.438±0.155 | 0.108±0.061 | 3.75×10−09 | 0.330 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bChromosome.
cNational Center for Biotechnology Information (NCBI) Database (Genome Build 37).
(B) Top 30 probes showing significant DNA hypomethylation in N samples of Cluster B1 compared to those of Cluster A (P < 0.05, Welch’s t test) and the largest differences in average DNA methylation levels between Clusters B1 and A (ΔβA–B1).
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean ± SD) | P | ΔβA–B1 | |
|---|---|---|---|---|---|---|---|
| Cluster A | Cluster B1 | ||||||
| cg24687051 | 6 | 73332073 | KCNQ5 | 0.543±0.108 | 0.059±0.051 | 1.31×10−16 | 0.484 |
| cg03168582 | 9 | 841850 | DMRT1 | 0.588±0.074 | 0.155±0.088 | 6.47×10−19 | 0.433 |
| cg17892556 | 19 | 12267464 | ZNF625 | 0.523±0.112 | 0.120±0.051 | 2.71×10−14 | 0.403 |
| cg07080358 | 2 | 68546507 | CNRIP1 | 0.542±0.092 | 0.142±0.067 | 2.37×10−17 | 0.400 |
| cg26309134 | 19 | 56879571 | ZNF542 | 0.519±0.092 | 0.121±0.048 | 1.41×10−16 | 0.398 |
| cg11657808 | 1 | 237205950 | RYR2 | 0.626±0.080 | 0.238±0.066 | 8.32×10−19 | 0.388 |
| cg11939071 | 12 | 113494429 | DTX1 | 0.465±0.070 | 0.084±0.053 | 1.89×10−20 | 0.381 |
| cg22029275 | 13 | 25745784 | AMER2 | 0.522±0.109 | 0.141±0.057 | 3.14×10−14 | 0.380 |
| cg22619018 | 8 | 4852624 | CSMD1 | 0.738±0.077 | 0.357±0.106 | 6.72×10−15 | 0.380 |
| cg12629325 | 5 | 140306458 | PCDHAC1 | 0.705±0.062 | 0.325±0.115 | 1.23×10−13 | 0.380 |
| cg17872757 | 11 | 128564180 | FLI1 | 0.439±0.135 | 0.066±0.034 | 5.48×10−11 | 0.373 |
| cg07017374 | 13 | 28674451 | FLT3 | 0.519±0.107 | 0.154±0.084 | 3.84×10−14 | 0.365 |
| cg13562911 | 6 | 11044106 | ELOVL2-AS1 | 0.501±0.101 | 0.139±0.060 | 9.84×10−15 | 0.362 |
| cg18671950 | 15 | 48936953 | FBN1 | 0.522±0.063 | 0.166±0.094 | 1.45×10−15 | 0.356 |
| cg06744574 | 1 | 49242359 | BEND5 | 0.417±0.150 | 0.065±0.064 | 5.20×10−10 | 0.352 |
| cg19118812 | 7 | 37488438 | ELMO1 | 0.417±0.137 | 0.066±0.046 | 1.41×10−10 | 0.350 |
| cg12874092 | 10 | 17271519 | VIM | 0.379±0.115 | 0.030±0.015 | 2.19×10−11 | 0.349 |
| cg09551147 | 10 | 106399957 | SORCS3 | 0.479±0.086 | 0.138±0.058 | 3.92×10−16 | 0.341 |
| cg25583174 | 4 | 123748386 | FGF2 | 0.439±0.112 | 0.099±0.064 | 8.84×10−13 | 0.340 |
| cg04034767 | 12 | 52400907 | GRASP | 0.453±0.136 | 0.114±0.046 | 2.58×10−10 | 0.338 |
| cg17525406 | 1 | 4715520 | AJAP1 | 0.709±0.068 | 0.372±0.111 | 6.16×10−13 | 0.337 |
| cg25886284 | 19 | 36909418 | ZFP82 | 0.459±0.084 | 0.122±0.075 | 8.20×10−16 | 0.336 |
| cg21475402 | 1 | 156612140 | BCAN | 0.569±0.074 | 0.232±0.059 | 7.45×10−18 | 0.336 |
| cg08383315 | 11 | 8190565 | RIC3 | 0.475±0.070 | 0.139±0.054 | 1.10×10−18 | 0.336 |
| cg21790626 | 19 | 58220494 | ZNF154 | 0.552±0.066 | 0.217±0.079 | 7.03×10−17 | 0.335 |
| cg16787600 | 10 | 106400880 | SORCS3 | 0.623±0.072 | 0.288±0.081 | 2.57×10−16 | 0.335 |
| cg20415809 | 2 | 182321855 | ITGA4 | 0.434±0.118 | 0.102±0.044 | 1.57×10−11 | 0.332 |
| cg10730712 | 5 | 178017827 | COL23A1 | 0.497±0.091 | 0.166±0.055 | 5.22×10−15 | 0.331 |
| cg01775414 | 22 | 45405404 | PHF21B | 0.496±0.096 | 0.165±0.062 | 2.47×10−14 | 0.330 |
| cg16778809 | 2 | 207308375 | ADAM23 | 0.438±0.155 | 0.108±0.061 | 3.75×10−09 | 0.330 |
| Probe IDa | Chb | Positionc | Gene symbol | DNA methylation levels (mean ± SD) | P | ΔβA–B1 | |
|---|---|---|---|---|---|---|---|
| Cluster A | Cluster B1 | ||||||
| cg24687051 | 6 | 73332073 | KCNQ5 | 0.543±0.108 | 0.059±0.051 | 1.31×10−16 | 0.484 |
| cg03168582 | 9 | 841850 | DMRT1 | 0.588±0.074 | 0.155±0.088 | 6.47×10−19 | 0.433 |
| cg17892556 | 19 | 12267464 | ZNF625 | 0.523±0.112 | 0.120±0.051 | 2.71×10−14 | 0.403 |
| cg07080358 | 2 | 68546507 | CNRIP1 | 0.542±0.092 | 0.142±0.067 | 2.37×10−17 | 0.400 |
| cg26309134 | 19 | 56879571 | ZNF542 | 0.519±0.092 | 0.121±0.048 | 1.41×10−16 | 0.398 |
| cg11657808 | 1 | 237205950 | RYR2 | 0.626±0.080 | 0.238±0.066 | 8.32×10−19 | 0.388 |
| cg11939071 | 12 | 113494429 | DTX1 | 0.465±0.070 | 0.084±0.053 | 1.89×10−20 | 0.381 |
| cg22029275 | 13 | 25745784 | AMER2 | 0.522±0.109 | 0.141±0.057 | 3.14×10−14 | 0.380 |
| cg22619018 | 8 | 4852624 | CSMD1 | 0.738±0.077 | 0.357±0.106 | 6.72×10−15 | 0.380 |
| cg12629325 | 5 | 140306458 | PCDHAC1 | 0.705±0.062 | 0.325±0.115 | 1.23×10−13 | 0.380 |
| cg17872757 | 11 | 128564180 | FLI1 | 0.439±0.135 | 0.066±0.034 | 5.48×10−11 | 0.373 |
| cg07017374 | 13 | 28674451 | FLT3 | 0.519±0.107 | 0.154±0.084 | 3.84×10−14 | 0.365 |
| cg13562911 | 6 | 11044106 | ELOVL2-AS1 | 0.501±0.101 | 0.139±0.060 | 9.84×10−15 | 0.362 |
| cg18671950 | 15 | 48936953 | FBN1 | 0.522±0.063 | 0.166±0.094 | 1.45×10−15 | 0.356 |
| cg06744574 | 1 | 49242359 | BEND5 | 0.417±0.150 | 0.065±0.064 | 5.20×10−10 | 0.352 |
| cg19118812 | 7 | 37488438 | ELMO1 | 0.417±0.137 | 0.066±0.046 | 1.41×10−10 | 0.350 |
| cg12874092 | 10 | 17271519 | VIM | 0.379±0.115 | 0.030±0.015 | 2.19×10−11 | 0.349 |
| cg09551147 | 10 | 106399957 | SORCS3 | 0.479±0.086 | 0.138±0.058 | 3.92×10−16 | 0.341 |
| cg25583174 | 4 | 123748386 | FGF2 | 0.439±0.112 | 0.099±0.064 | 8.84×10−13 | 0.340 |
| cg04034767 | 12 | 52400907 | GRASP | 0.453±0.136 | 0.114±0.046 | 2.58×10−10 | 0.338 |
| cg17525406 | 1 | 4715520 | AJAP1 | 0.709±0.068 | 0.372±0.111 | 6.16×10−13 | 0.337 |
| cg25886284 | 19 | 36909418 | ZFP82 | 0.459±0.084 | 0.122±0.075 | 8.20×10−16 | 0.336 |
| cg21475402 | 1 | 156612140 | BCAN | 0.569±0.074 | 0.232±0.059 | 7.45×10−18 | 0.336 |
| cg08383315 | 11 | 8190565 | RIC3 | 0.475±0.070 | 0.139±0.054 | 1.10×10−18 | 0.336 |
| cg21790626 | 19 | 58220494 | ZNF154 | 0.552±0.066 | 0.217±0.079 | 7.03×10−17 | 0.335 |
| cg16787600 | 10 | 106400880 | SORCS3 | 0.623±0.072 | 0.288±0.081 | 2.57×10−16 | 0.335 |
| cg20415809 | 2 | 182321855 | ITGA4 | 0.434±0.118 | 0.102±0.044 | 1.57×10−11 | 0.332 |
| cg10730712 | 5 | 178017827 | COL23A1 | 0.497±0.091 | 0.166±0.055 | 5.22×10−15 | 0.331 |
| cg01775414 | 22 | 45405404 | PHF21B | 0.496±0.096 | 0.165±0.062 | 2.47×10−14 | 0.330 |
| cg16778809 | 2 | 207308375 | ADAM23 | 0.438±0.155 | 0.108±0.061 | 3.75×10−09 | 0.330 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bChromosome.
cNational Center for Biotechnology Information (NCBI) Database (Genome Build 37).
Since multiple probes around the transcription start site of a gene are incorporated in the Infinium HumanMethylation27 Bead Array, DNA methylation levels of all probes for the same genes other than those included in Table 2 have been summarized in Supplementary Table 5 (available at Carcinogenesis Online). For 59 genes out of the 60 listed in Table 2, the average DNA methylation levels for all probes, including probes other than those included in Table 2 for the same genes, again showed statistically significant differences between Clusters A and B1, indicating that the probes listed in Table 2 well represented the DNA methylation status around the transcription start sites of the genes.
Impact of DNA methylation levels of probes characterizing the epigenetic clustering in T samples on tumor aggressiveness and patient outcome
In order to examine whether DNA methylation profiles in N samples characterizing the epigenetic clustering were inherited by gastric carcinomas themselves, we focused on DNA methylation levels in T samples (βT) for the identified top 60 probes. In T samples (βT), DNA methylation levels for 19 genes included in Table 2A and 5 genes included in Table 2B (24 genes in total) were again significantly correlated with an undifferentiated histological type, deeper invasion and/or a higher pathological TNM stage (Table 3).
Correlations between DNA methylation levels of probes characterizing the epigenetic clustering in cancerous tissue samples and clinicopathological parameters of gastric cancers
| Target ID a | Gene symbol | Predominant histological classificationb | Most aggressive histological classificationc | Tumor stage | Pathological Tumor-Node-Metastasis stage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average βTd | P valueh | Average βTd | P valuei | Average βTd | P valuei | Average βTd | P valueh | ||||||||
| Diff.e | Undiff.f | Mucing | Diff.e | Undiff.f | pT1–pT2 | pT3–pT4 | IA–IB | IIA–IIB | IIIA–IV | ||||||
| (A) Probes listed in Table 2A | |||||||||||||||
| cg23743114 | CCL15−CCL14 | 0.398 | 0.460 | 0.351 | 7.71×10−2 | 0.372 | 0.443 | 2.07×10−2 | 0.430 | 0.422 | 8.12×10−1 | 0.423 | 0.430 | 0.423 | 9.82×10−1 |
| cg02192965 | SLC3A1 | 0.406 | 0.516 | 0.425 | 2.63×10−3 | 0.381 | 0.483 | 2.08×10−2 | 0.425 | 0.465 | 3.01×10−1 | 0.411 | 0.497 | 0.459 | 2.56×10−1 |
| cg14934821 | GPSM1 | 0.531 | 0.566 | 0.482 | 4.65×10−1 | 0.535 | 0.549 | 7.28×10−1 | 0.458 | 0.571 | 6.91×10−4 | 0.467 | 0.548 | 0.571 | 4.28×10−2 |
| cg07220939 | SLC22A12 | 0.316 | 0.418 | 0.413 | 2.00×10−2 | 0.296 | 0.389 | 1.81×10−2 | 0.374 | 0.362 | 7.97×10−1 | 0.357 | 0.429 | 0.349 | 2.73×10−1 |
| cg26530341 | LOC389641 | 0.392 | 0.450 | 0.388 | 9.32×10−2 | 0.375 | 0.433 | 4.68×10−2 | 0.365 | 0.433 | 1.97×10−2 | 0.370 | 0.423 | 0.432 | 1.76×10−1 |
| cg04968426 | PPP1R14D | 0.625 | 0.714 | 0.666 | 2.52×10−2 | 0.590 | 0.694 | 9.07×10−3 | 0.632 | 0.676 | 2.69×10−1 | 0.618 | 0.677 | 0.679 | 3.31×10−1 |
| cg03545635 | CHST12 | 0.712 | 0.734 | 0.682 | 4.51×10−1 | 0.712 | 0.725 | 5.58×10−1 | 0.681 | 0.733 | 3.55×10−2 | 0.680 | 0.735 | 0.731 | 1.06×10−1 |
| cg07150830 | NOS2 | 0.670 | 0.779 | 0.629 | 5.39×10−3 | 0.661 | 0.738 | 3.79×10−2 | 0.691 | 0.725 | 4.00×10−1 | 0.687 | 0.659 | 0.744 | 1.37×10−1 |
| cg21122774 | SARDH | 0.528 | 0.633 | 0.492 | 4.43×10−3 | 0.523 | 0.593 | 4.29×10−2 | 0.508 | 0.593 | 1.70×10−2 | 0.496 | 0.605 | 0.592 | 4.51×10−2 |
| cg17778120 | RBP2 | 0.589 | 0.670 | 0.699 | 2.89×10−2 | 0.596 | 0.640 | 2.07×10−1 | 0.611 | 0.634 | 5.29×10−1 | 0.606 | 0.620 | 0.638 | 7.02×10−1 |
| cg09081544 | MUC13 | 0.448 | 0.526 | 0.443 | 4.16×10−2 | 0.443 | 0.497 | 1.11×10−1 | 0.463 | 0.489 | 4.63×10−1 | 0.455 | 0.501 | 0.487 | 6.35×10−1 |
| cg09448875 | ABCC2 | 0.423 | 0.512 | 0.428 | 1.34×10−2 | 0.419 | 0.479 | 7.36×10−2 | 0.450 | 0.467 | 6.19×10−1 | 0.441 | 0.490 | 0.464 | 6.15×10−1 |
| cg03077492 | CCL28 | 0.773 | 0.817 | 0.736 | 8.63×10−2 | 0.764 | 0.802 | 1.56×10−1 | 0.744 | 0.806 | 3.49×10−2 | 0.752 | 0.792 | 0.805 | 1.42×10−1 |
| cg24027679 | SLC2A7 | 0.630 | 0.696 | 0.631 | 9.85×10−2 | 0.629 | 0.671 | 2.42×10−1 | 0.604 | 0.676 | 4.02×10−2 | 0.587 | 0.692 | 0.675 | 4.43×10−2 |
| cg20683151 | TM4SF20 | 0.591 | 0.668 | 0.570 | 3.74×10−2 | 0.574 | 0.643 | 5.37×10−2 | 0.624 | 0.625 | 9.87×10−1 | 0.619 | 0.650 | 0.620 | 7.70×10−1 |
| cg21591452 | TMEM105 | 0.683 | 0.777 | 0.730 | 8.61×10−3 | 0.649 | 0.755 | 3.12×10−3 | 0.721 | 0.728 | 8.55×10−1 | 0.713 | 0.733 | 0.730 | 8.91×10−1 |
| cg06277277 | NR1I3 | 0.512 | 0.625 | 0.565 | 7.81×10−4 | 0.525 | 0.578 | 1.27×10−1 | 0.570 | 0.562 | 8.13×10−1 | 0.565 | 0.547 | 0.568 | 8.76×10−1 |
| cg11920519 | MAP1LC3A | 0.562 | 0.661 | 0.530 | 1.31×10−2 | 0.552 | 0.625 | 6.41×10−2 | 0.602 | 0.607 | 9.16×10−1 | 0.594 | 0.611 | 0.608 | 9.43×10−1 |
| cg16575408 | MMP1 | 0.682 | 0.760 | 0.620 | 3.56×10−2 | 0.643 | 0.741 | 1.01×10−2 | 0.677 | 0.726 | 2.58×10−1 | 0.673 | 0.708 | 0.731 | 3.64×10−1 |
| (B) Probes listed in Table 2B | |||||||||||||||
| cg11657808 | RYR2 | 0.686 | 0.618 | 0.636 | 1.27×10−2 | 0.709 | 0.634 | 2.91×10−2 | 0.692 | 0.643 | 1.32×10−1 | 0.686 | 0.621 | 0.652 | 4.83×10−1 |
| cg11939071 | DTX1 | 0.328 | 0.409 | 0.517 | 1.08×10−2 | 0.280 | 0.402 | 1.69×10−2 | 0.452 | 0.345 | 2.69×10−2 | 0.442 | 0.353 | 0.350 | 2.50×10−1 |
| cg22619018 | CSMD1 | 0.576 | 0.632 | 0.638 | 5.39×10−2 | 0.572 | 0.614 | 4.76×10−1 | 0.701 | 0.574 | 2.30×10−2 | 0.698 | 0.519 | 0.594 | 8.28×10−2 |
| cg18671950 | FBN1 | 0.511 | 0.466 | 0.594 | 3.02×10−2 | 0.533 | 0.479 | 1.61×10−1 | 0.554 | 0.475 | 1.31×10−2 | 0.553 | 0.452 | 0.484 | 1.87×10−1 |
| cg08383315 | RIC3 | 0.459 | 0.426 | 0.505 | 5.64×10−2 | 0.479 | 0.433 | 2.47×10−1 | 0.517 | 0.424 | 1.25×10−2 | 0.510 | 0.401 | 0.436 | 1.34×10−1 |
| Target ID a | Gene symbol | Predominant histological classificationb | Most aggressive histological classificationc | Tumor stage | Pathological Tumor-Node-Metastasis stage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average βTd | P valueh | Average βTd | P valuei | Average βTd | P valuei | Average βTd | P valueh | ||||||||
| Diff.e | Undiff.f | Mucing | Diff.e | Undiff.f | pT1–pT2 | pT3–pT4 | IA–IB | IIA–IIB | IIIA–IV | ||||||
| (A) Probes listed in Table 2A | |||||||||||||||
| cg23743114 | CCL15−CCL14 | 0.398 | 0.460 | 0.351 | 7.71×10−2 | 0.372 | 0.443 | 2.07×10−2 | 0.430 | 0.422 | 8.12×10−1 | 0.423 | 0.430 | 0.423 | 9.82×10−1 |
| cg02192965 | SLC3A1 | 0.406 | 0.516 | 0.425 | 2.63×10−3 | 0.381 | 0.483 | 2.08×10−2 | 0.425 | 0.465 | 3.01×10−1 | 0.411 | 0.497 | 0.459 | 2.56×10−1 |
| cg14934821 | GPSM1 | 0.531 | 0.566 | 0.482 | 4.65×10−1 | 0.535 | 0.549 | 7.28×10−1 | 0.458 | 0.571 | 6.91×10−4 | 0.467 | 0.548 | 0.571 | 4.28×10−2 |
| cg07220939 | SLC22A12 | 0.316 | 0.418 | 0.413 | 2.00×10−2 | 0.296 | 0.389 | 1.81×10−2 | 0.374 | 0.362 | 7.97×10−1 | 0.357 | 0.429 | 0.349 | 2.73×10−1 |
| cg26530341 | LOC389641 | 0.392 | 0.450 | 0.388 | 9.32×10−2 | 0.375 | 0.433 | 4.68×10−2 | 0.365 | 0.433 | 1.97×10−2 | 0.370 | 0.423 | 0.432 | 1.76×10−1 |
| cg04968426 | PPP1R14D | 0.625 | 0.714 | 0.666 | 2.52×10−2 | 0.590 | 0.694 | 9.07×10−3 | 0.632 | 0.676 | 2.69×10−1 | 0.618 | 0.677 | 0.679 | 3.31×10−1 |
| cg03545635 | CHST12 | 0.712 | 0.734 | 0.682 | 4.51×10−1 | 0.712 | 0.725 | 5.58×10−1 | 0.681 | 0.733 | 3.55×10−2 | 0.680 | 0.735 | 0.731 | 1.06×10−1 |
| cg07150830 | NOS2 | 0.670 | 0.779 | 0.629 | 5.39×10−3 | 0.661 | 0.738 | 3.79×10−2 | 0.691 | 0.725 | 4.00×10−1 | 0.687 | 0.659 | 0.744 | 1.37×10−1 |
| cg21122774 | SARDH | 0.528 | 0.633 | 0.492 | 4.43×10−3 | 0.523 | 0.593 | 4.29×10−2 | 0.508 | 0.593 | 1.70×10−2 | 0.496 | 0.605 | 0.592 | 4.51×10−2 |
| cg17778120 | RBP2 | 0.589 | 0.670 | 0.699 | 2.89×10−2 | 0.596 | 0.640 | 2.07×10−1 | 0.611 | 0.634 | 5.29×10−1 | 0.606 | 0.620 | 0.638 | 7.02×10−1 |
| cg09081544 | MUC13 | 0.448 | 0.526 | 0.443 | 4.16×10−2 | 0.443 | 0.497 | 1.11×10−1 | 0.463 | 0.489 | 4.63×10−1 | 0.455 | 0.501 | 0.487 | 6.35×10−1 |
| cg09448875 | ABCC2 | 0.423 | 0.512 | 0.428 | 1.34×10−2 | 0.419 | 0.479 | 7.36×10−2 | 0.450 | 0.467 | 6.19×10−1 | 0.441 | 0.490 | 0.464 | 6.15×10−1 |
| cg03077492 | CCL28 | 0.773 | 0.817 | 0.736 | 8.63×10−2 | 0.764 | 0.802 | 1.56×10−1 | 0.744 | 0.806 | 3.49×10−2 | 0.752 | 0.792 | 0.805 | 1.42×10−1 |
| cg24027679 | SLC2A7 | 0.630 | 0.696 | 0.631 | 9.85×10−2 | 0.629 | 0.671 | 2.42×10−1 | 0.604 | 0.676 | 4.02×10−2 | 0.587 | 0.692 | 0.675 | 4.43×10−2 |
| cg20683151 | TM4SF20 | 0.591 | 0.668 | 0.570 | 3.74×10−2 | 0.574 | 0.643 | 5.37×10−2 | 0.624 | 0.625 | 9.87×10−1 | 0.619 | 0.650 | 0.620 | 7.70×10−1 |
| cg21591452 | TMEM105 | 0.683 | 0.777 | 0.730 | 8.61×10−3 | 0.649 | 0.755 | 3.12×10−3 | 0.721 | 0.728 | 8.55×10−1 | 0.713 | 0.733 | 0.730 | 8.91×10−1 |
| cg06277277 | NR1I3 | 0.512 | 0.625 | 0.565 | 7.81×10−4 | 0.525 | 0.578 | 1.27×10−1 | 0.570 | 0.562 | 8.13×10−1 | 0.565 | 0.547 | 0.568 | 8.76×10−1 |
| cg11920519 | MAP1LC3A | 0.562 | 0.661 | 0.530 | 1.31×10−2 | 0.552 | 0.625 | 6.41×10−2 | 0.602 | 0.607 | 9.16×10−1 | 0.594 | 0.611 | 0.608 | 9.43×10−1 |
| cg16575408 | MMP1 | 0.682 | 0.760 | 0.620 | 3.56×10−2 | 0.643 | 0.741 | 1.01×10−2 | 0.677 | 0.726 | 2.58×10−1 | 0.673 | 0.708 | 0.731 | 3.64×10−1 |
| (B) Probes listed in Table 2B | |||||||||||||||
| cg11657808 | RYR2 | 0.686 | 0.618 | 0.636 | 1.27×10−2 | 0.709 | 0.634 | 2.91×10−2 | 0.692 | 0.643 | 1.32×10−1 | 0.686 | 0.621 | 0.652 | 4.83×10−1 |
| cg11939071 | DTX1 | 0.328 | 0.409 | 0.517 | 1.08×10−2 | 0.280 | 0.402 | 1.69×10−2 | 0.452 | 0.345 | 2.69×10−2 | 0.442 | 0.353 | 0.350 | 2.50×10−1 |
| cg22619018 | CSMD1 | 0.576 | 0.632 | 0.638 | 5.39×10−2 | 0.572 | 0.614 | 4.76×10−1 | 0.701 | 0.574 | 2.30×10−2 | 0.698 | 0.519 | 0.594 | 8.28×10−2 |
| cg18671950 | FBN1 | 0.511 | 0.466 | 0.594 | 3.02×10−2 | 0.533 | 0.479 | 1.61×10−1 | 0.554 | 0.475 | 1.31×10−2 | 0.553 | 0.452 | 0.484 | 1.87×10−1 |
| cg08383315 | RIC3 | 0.459 | 0.426 | 0.505 | 5.64×10−2 | 0.479 | 0.433 | 2.47×10−1 | 0.517 | 0.424 | 1.25×10−2 | 0.510 | 0.401 | 0.436 | 1.34×10−1 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bIf the tumor showed heterogeneity, findings in the predominant area were described.
cIf the tumor showed heterogeneity, the most aggressive features of the tumor were described.
dAverage DNA methylation levels in T samples.
eDifferentiated. fUndifferentiated. gMucin-producing.
hP values (ANOVA) and iWelch’s t test (P values of < 0.05 are underlined).
Correlations between DNA methylation levels of probes characterizing the epigenetic clustering in cancerous tissue samples and clinicopathological parameters of gastric cancers
| Target ID a | Gene symbol | Predominant histological classificationb | Most aggressive histological classificationc | Tumor stage | Pathological Tumor-Node-Metastasis stage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average βTd | P valueh | Average βTd | P valuei | Average βTd | P valuei | Average βTd | P valueh | ||||||||
| Diff.e | Undiff.f | Mucing | Diff.e | Undiff.f | pT1–pT2 | pT3–pT4 | IA–IB | IIA–IIB | IIIA–IV | ||||||
| (A) Probes listed in Table 2A | |||||||||||||||
| cg23743114 | CCL15−CCL14 | 0.398 | 0.460 | 0.351 | 7.71×10−2 | 0.372 | 0.443 | 2.07×10−2 | 0.430 | 0.422 | 8.12×10−1 | 0.423 | 0.430 | 0.423 | 9.82×10−1 |
| cg02192965 | SLC3A1 | 0.406 | 0.516 | 0.425 | 2.63×10−3 | 0.381 | 0.483 | 2.08×10−2 | 0.425 | 0.465 | 3.01×10−1 | 0.411 | 0.497 | 0.459 | 2.56×10−1 |
| cg14934821 | GPSM1 | 0.531 | 0.566 | 0.482 | 4.65×10−1 | 0.535 | 0.549 | 7.28×10−1 | 0.458 | 0.571 | 6.91×10−4 | 0.467 | 0.548 | 0.571 | 4.28×10−2 |
| cg07220939 | SLC22A12 | 0.316 | 0.418 | 0.413 | 2.00×10−2 | 0.296 | 0.389 | 1.81×10−2 | 0.374 | 0.362 | 7.97×10−1 | 0.357 | 0.429 | 0.349 | 2.73×10−1 |
| cg26530341 | LOC389641 | 0.392 | 0.450 | 0.388 | 9.32×10−2 | 0.375 | 0.433 | 4.68×10−2 | 0.365 | 0.433 | 1.97×10−2 | 0.370 | 0.423 | 0.432 | 1.76×10−1 |
| cg04968426 | PPP1R14D | 0.625 | 0.714 | 0.666 | 2.52×10−2 | 0.590 | 0.694 | 9.07×10−3 | 0.632 | 0.676 | 2.69×10−1 | 0.618 | 0.677 | 0.679 | 3.31×10−1 |
| cg03545635 | CHST12 | 0.712 | 0.734 | 0.682 | 4.51×10−1 | 0.712 | 0.725 | 5.58×10−1 | 0.681 | 0.733 | 3.55×10−2 | 0.680 | 0.735 | 0.731 | 1.06×10−1 |
| cg07150830 | NOS2 | 0.670 | 0.779 | 0.629 | 5.39×10−3 | 0.661 | 0.738 | 3.79×10−2 | 0.691 | 0.725 | 4.00×10−1 | 0.687 | 0.659 | 0.744 | 1.37×10−1 |
| cg21122774 | SARDH | 0.528 | 0.633 | 0.492 | 4.43×10−3 | 0.523 | 0.593 | 4.29×10−2 | 0.508 | 0.593 | 1.70×10−2 | 0.496 | 0.605 | 0.592 | 4.51×10−2 |
| cg17778120 | RBP2 | 0.589 | 0.670 | 0.699 | 2.89×10−2 | 0.596 | 0.640 | 2.07×10−1 | 0.611 | 0.634 | 5.29×10−1 | 0.606 | 0.620 | 0.638 | 7.02×10−1 |
| cg09081544 | MUC13 | 0.448 | 0.526 | 0.443 | 4.16×10−2 | 0.443 | 0.497 | 1.11×10−1 | 0.463 | 0.489 | 4.63×10−1 | 0.455 | 0.501 | 0.487 | 6.35×10−1 |
| cg09448875 | ABCC2 | 0.423 | 0.512 | 0.428 | 1.34×10−2 | 0.419 | 0.479 | 7.36×10−2 | 0.450 | 0.467 | 6.19×10−1 | 0.441 | 0.490 | 0.464 | 6.15×10−1 |
| cg03077492 | CCL28 | 0.773 | 0.817 | 0.736 | 8.63×10−2 | 0.764 | 0.802 | 1.56×10−1 | 0.744 | 0.806 | 3.49×10−2 | 0.752 | 0.792 | 0.805 | 1.42×10−1 |
| cg24027679 | SLC2A7 | 0.630 | 0.696 | 0.631 | 9.85×10−2 | 0.629 | 0.671 | 2.42×10−1 | 0.604 | 0.676 | 4.02×10−2 | 0.587 | 0.692 | 0.675 | 4.43×10−2 |
| cg20683151 | TM4SF20 | 0.591 | 0.668 | 0.570 | 3.74×10−2 | 0.574 | 0.643 | 5.37×10−2 | 0.624 | 0.625 | 9.87×10−1 | 0.619 | 0.650 | 0.620 | 7.70×10−1 |
| cg21591452 | TMEM105 | 0.683 | 0.777 | 0.730 | 8.61×10−3 | 0.649 | 0.755 | 3.12×10−3 | 0.721 | 0.728 | 8.55×10−1 | 0.713 | 0.733 | 0.730 | 8.91×10−1 |
| cg06277277 | NR1I3 | 0.512 | 0.625 | 0.565 | 7.81×10−4 | 0.525 | 0.578 | 1.27×10−1 | 0.570 | 0.562 | 8.13×10−1 | 0.565 | 0.547 | 0.568 | 8.76×10−1 |
| cg11920519 | MAP1LC3A | 0.562 | 0.661 | 0.530 | 1.31×10−2 | 0.552 | 0.625 | 6.41×10−2 | 0.602 | 0.607 | 9.16×10−1 | 0.594 | 0.611 | 0.608 | 9.43×10−1 |
| cg16575408 | MMP1 | 0.682 | 0.760 | 0.620 | 3.56×10−2 | 0.643 | 0.741 | 1.01×10−2 | 0.677 | 0.726 | 2.58×10−1 | 0.673 | 0.708 | 0.731 | 3.64×10−1 |
| (B) Probes listed in Table 2B | |||||||||||||||
| cg11657808 | RYR2 | 0.686 | 0.618 | 0.636 | 1.27×10−2 | 0.709 | 0.634 | 2.91×10−2 | 0.692 | 0.643 | 1.32×10−1 | 0.686 | 0.621 | 0.652 | 4.83×10−1 |
| cg11939071 | DTX1 | 0.328 | 0.409 | 0.517 | 1.08×10−2 | 0.280 | 0.402 | 1.69×10−2 | 0.452 | 0.345 | 2.69×10−2 | 0.442 | 0.353 | 0.350 | 2.50×10−1 |
| cg22619018 | CSMD1 | 0.576 | 0.632 | 0.638 | 5.39×10−2 | 0.572 | 0.614 | 4.76×10−1 | 0.701 | 0.574 | 2.30×10−2 | 0.698 | 0.519 | 0.594 | 8.28×10−2 |
| cg18671950 | FBN1 | 0.511 | 0.466 | 0.594 | 3.02×10−2 | 0.533 | 0.479 | 1.61×10−1 | 0.554 | 0.475 | 1.31×10−2 | 0.553 | 0.452 | 0.484 | 1.87×10−1 |
| cg08383315 | RIC3 | 0.459 | 0.426 | 0.505 | 5.64×10−2 | 0.479 | 0.433 | 2.47×10−1 | 0.517 | 0.424 | 1.25×10−2 | 0.510 | 0.401 | 0.436 | 1.34×10−1 |
| Target ID a | Gene symbol | Predominant histological classificationb | Most aggressive histological classificationc | Tumor stage | Pathological Tumor-Node-Metastasis stage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average βTd | P valueh | Average βTd | P valuei | Average βTd | P valuei | Average βTd | P valueh | ||||||||
| Diff.e | Undiff.f | Mucing | Diff.e | Undiff.f | pT1–pT2 | pT3–pT4 | IA–IB | IIA–IIB | IIIA–IV | ||||||
| (A) Probes listed in Table 2A | |||||||||||||||
| cg23743114 | CCL15−CCL14 | 0.398 | 0.460 | 0.351 | 7.71×10−2 | 0.372 | 0.443 | 2.07×10−2 | 0.430 | 0.422 | 8.12×10−1 | 0.423 | 0.430 | 0.423 | 9.82×10−1 |
| cg02192965 | SLC3A1 | 0.406 | 0.516 | 0.425 | 2.63×10−3 | 0.381 | 0.483 | 2.08×10−2 | 0.425 | 0.465 | 3.01×10−1 | 0.411 | 0.497 | 0.459 | 2.56×10−1 |
| cg14934821 | GPSM1 | 0.531 | 0.566 | 0.482 | 4.65×10−1 | 0.535 | 0.549 | 7.28×10−1 | 0.458 | 0.571 | 6.91×10−4 | 0.467 | 0.548 | 0.571 | 4.28×10−2 |
| cg07220939 | SLC22A12 | 0.316 | 0.418 | 0.413 | 2.00×10−2 | 0.296 | 0.389 | 1.81×10−2 | 0.374 | 0.362 | 7.97×10−1 | 0.357 | 0.429 | 0.349 | 2.73×10−1 |
| cg26530341 | LOC389641 | 0.392 | 0.450 | 0.388 | 9.32×10−2 | 0.375 | 0.433 | 4.68×10−2 | 0.365 | 0.433 | 1.97×10−2 | 0.370 | 0.423 | 0.432 | 1.76×10−1 |
| cg04968426 | PPP1R14D | 0.625 | 0.714 | 0.666 | 2.52×10−2 | 0.590 | 0.694 | 9.07×10−3 | 0.632 | 0.676 | 2.69×10−1 | 0.618 | 0.677 | 0.679 | 3.31×10−1 |
| cg03545635 | CHST12 | 0.712 | 0.734 | 0.682 | 4.51×10−1 | 0.712 | 0.725 | 5.58×10−1 | 0.681 | 0.733 | 3.55×10−2 | 0.680 | 0.735 | 0.731 | 1.06×10−1 |
| cg07150830 | NOS2 | 0.670 | 0.779 | 0.629 | 5.39×10−3 | 0.661 | 0.738 | 3.79×10−2 | 0.691 | 0.725 | 4.00×10−1 | 0.687 | 0.659 | 0.744 | 1.37×10−1 |
| cg21122774 | SARDH | 0.528 | 0.633 | 0.492 | 4.43×10−3 | 0.523 | 0.593 | 4.29×10−2 | 0.508 | 0.593 | 1.70×10−2 | 0.496 | 0.605 | 0.592 | 4.51×10−2 |
| cg17778120 | RBP2 | 0.589 | 0.670 | 0.699 | 2.89×10−2 | 0.596 | 0.640 | 2.07×10−1 | 0.611 | 0.634 | 5.29×10−1 | 0.606 | 0.620 | 0.638 | 7.02×10−1 |
| cg09081544 | MUC13 | 0.448 | 0.526 | 0.443 | 4.16×10−2 | 0.443 | 0.497 | 1.11×10−1 | 0.463 | 0.489 | 4.63×10−1 | 0.455 | 0.501 | 0.487 | 6.35×10−1 |
| cg09448875 | ABCC2 | 0.423 | 0.512 | 0.428 | 1.34×10−2 | 0.419 | 0.479 | 7.36×10−2 | 0.450 | 0.467 | 6.19×10−1 | 0.441 | 0.490 | 0.464 | 6.15×10−1 |
| cg03077492 | CCL28 | 0.773 | 0.817 | 0.736 | 8.63×10−2 | 0.764 | 0.802 | 1.56×10−1 | 0.744 | 0.806 | 3.49×10−2 | 0.752 | 0.792 | 0.805 | 1.42×10−1 |
| cg24027679 | SLC2A7 | 0.630 | 0.696 | 0.631 | 9.85×10−2 | 0.629 | 0.671 | 2.42×10−1 | 0.604 | 0.676 | 4.02×10−2 | 0.587 | 0.692 | 0.675 | 4.43×10−2 |
| cg20683151 | TM4SF20 | 0.591 | 0.668 | 0.570 | 3.74×10−2 | 0.574 | 0.643 | 5.37×10−2 | 0.624 | 0.625 | 9.87×10−1 | 0.619 | 0.650 | 0.620 | 7.70×10−1 |
| cg21591452 | TMEM105 | 0.683 | 0.777 | 0.730 | 8.61×10−3 | 0.649 | 0.755 | 3.12×10−3 | 0.721 | 0.728 | 8.55×10−1 | 0.713 | 0.733 | 0.730 | 8.91×10−1 |
| cg06277277 | NR1I3 | 0.512 | 0.625 | 0.565 | 7.81×10−4 | 0.525 | 0.578 | 1.27×10−1 | 0.570 | 0.562 | 8.13×10−1 | 0.565 | 0.547 | 0.568 | 8.76×10−1 |
| cg11920519 | MAP1LC3A | 0.562 | 0.661 | 0.530 | 1.31×10−2 | 0.552 | 0.625 | 6.41×10−2 | 0.602 | 0.607 | 9.16×10−1 | 0.594 | 0.611 | 0.608 | 9.43×10−1 |
| cg16575408 | MMP1 | 0.682 | 0.760 | 0.620 | 3.56×10−2 | 0.643 | 0.741 | 1.01×10−2 | 0.677 | 0.726 | 2.58×10−1 | 0.673 | 0.708 | 0.731 | 3.64×10−1 |
| (B) Probes listed in Table 2B | |||||||||||||||
| cg11657808 | RYR2 | 0.686 | 0.618 | 0.636 | 1.27×10−2 | 0.709 | 0.634 | 2.91×10−2 | 0.692 | 0.643 | 1.32×10−1 | 0.686 | 0.621 | 0.652 | 4.83×10−1 |
| cg11939071 | DTX1 | 0.328 | 0.409 | 0.517 | 1.08×10−2 | 0.280 | 0.402 | 1.69×10−2 | 0.452 | 0.345 | 2.69×10−2 | 0.442 | 0.353 | 0.350 | 2.50×10−1 |
| cg22619018 | CSMD1 | 0.576 | 0.632 | 0.638 | 5.39×10−2 | 0.572 | 0.614 | 4.76×10−1 | 0.701 | 0.574 | 2.30×10−2 | 0.698 | 0.519 | 0.594 | 8.28×10−2 |
| cg18671950 | FBN1 | 0.511 | 0.466 | 0.594 | 3.02×10−2 | 0.533 | 0.479 | 1.61×10−1 | 0.554 | 0.475 | 1.31×10−2 | 0.553 | 0.452 | 0.484 | 1.87×10−1 |
| cg08383315 | RIC3 | 0.459 | 0.426 | 0.505 | 5.64×10−2 | 0.479 | 0.433 | 2.47×10−1 | 0.517 | 0.424 | 1.25×10−2 | 0.510 | 0.401 | 0.436 | 1.34×10−1 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bIf the tumor showed heterogeneity, findings in the predominant area were described.
cIf the tumor showed heterogeneity, the most aggressive features of the tumor were described.
dAverage DNA methylation levels in T samples.
eDifferentiated. fUndifferentiated. gMucin-producing.
hP values (ANOVA) and iWelch’s t test (P values of < 0.05 are underlined).
In order to examine the prognostic impact of DNA methylation levels in T samples (βT) for the top 60 identified probes, ROC curves were generated. The Youden index for each probe was used as a cut-off value when examining correlations between DNA methylation levels (βT) and patient outcome (Supplementary Table 4, available at Carcinogenesis Online). For each of the 60 probes, survival curves for patients belonging to groups with higher (βT ≥ Youden index) and lower (βT < Youden index) DNA methylation levels were generated by the Kaplan–Meier method. For 12 genes included in Table 2A, DNA methylation levels in T samples (βT) were significantly correlated with cancer recurrence in the 86 patients from whom the samples had been obtained, and who underwent complete resection. For 14 genes included in Table 2A, DNA methylation levels in T samples (βT) were significantly correlated with disease-related death in all of the 105 patients from whom the samples had been obtained. P values for the 12 and 14 probes (17 in total) determined by the log-rank test are summarized in Supplementary Table 4 (available at Carcinogenesis Online), and the representative 10 Kaplan–Meier curves showing the recurrence-free or overall survival rates with the smallest P values are shown in Figure 2A.
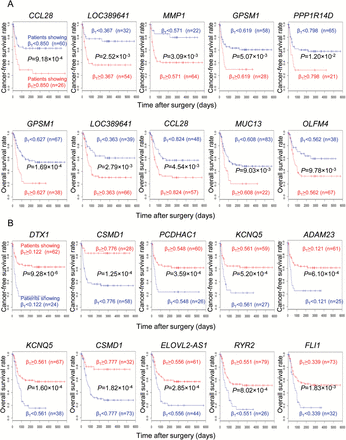
Prognostic impact of DNA methylation levels in tumorous tissue (T) samples for hallmark genes characterizing the epigenetic clustering based on DNA methylation profiles at the precancerous stage. (A) Kaplan–Meier survival curves for patients showing higher (βT ≥ Youden index) and lower (βT < Youden index) DNA methylation levels in T samples for genes listed in Table 2A. Representative genes (CCL28, LOC389641, MMP1, GPSM1 and PPP1R14D) showing the smallest P values for the recurrence-free survival rate of the 86 patients who underwent complete resection, and representative genes (GPSM1, LOC389641, CCL28, MUC13 and OLFM4) showing the smallest P values for the overall survival rate of all 105 patients, are shown. (B) Kaplan–Meier survival curves for patients showing higher (βT ≥ Youden index) and lower (βT < Youden index) DNA methylation levels in T samples for genes listed in Table 2B. Representative genes (DTX1, CSMD1, PCDHAC1, KCNQ5 and ADAM23) showing the smallest P values for the recurrence-free survival rate of the 86 patients who underwent complete resection, and representative genes (KCNQ5, CSMD1, ELOVL2-AS1, RYR2 and FLI1) showing the smallest P values for the overall survival rate of all 105 patients, are shown.
For 21 genes included in Table 2B, DNA methylation levels in T samples (βT) were significantly correlated with cancer recurrence in the 86 patients from whom the samples had been obtained, and who underwent complete resection. For 21 genes included in Table 2B, DNA methylation levels in T samples (βT) were significantly correlated with disease-related death in all of the 105 patients from whom the samples had been obtained. P values for the 21 and 21 probes (24 in total) obtained by the log-rank test are summarized in Supplementary Table 6 (available at Carcinogenesis Online), and the representative 10 Kaplan–Meier curves showing the recurrence-free or overall survival rates with the smallest P values are shown in Figure 2B.
Multivariate analyses using the Cox proportional hazards regression model revealed that DNA methylation levels in T samples (βT) in 11 and 7 genes (13 in total) included in Table 2 were significant prognostic factors (for recurrence and disease-related death, respectively), being independent of histological differentiation, depth of invasion and pathological TNM stage (Table 4).
Multivariate analyses using the Cox proportional hazards regression model for recurrence and disease-related death of patients with gastric cancers
| Probe IDa | Gene symbol | Recurrenceb | Disease-related deathb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | ||
| cg24687051 | KCNQ5 | 4.75×10−2 | 0.587 | 0.647 | 0.846 | 4.52×10−3 | 0.177 | 0.613 | 0.384 | 0.815 | 6.25×10−4 |
| cg03168582 | DMRT1 | 1.50×10−4 | 0.774 | 0.612 | 0.961 | 1.71×10−3 | 6.05×10−3 | 0.841 | 0.356 | 0.910 | 3.33×10−4 |
| cg07080358 | CNRIP1 | 3.64×10−2 | 0.444 | 0.670 | 0.842 | 2.29×10−3 | 7.49×10−2 | 0.501 | 0.458 | 0.816 | 3.41×10−4 |
| cg11939071 | DTX1 | 2.37×10−3 | 0.672 | 0.141 | 0.824 | 2.18×10−3 | 5.37×10−2 | 0.662 | 0.126 | 0.941 | 4.25×10−4 |
| cg22029275 | AMER2 | 5.30×10−2 | 0.270 | 0.328 | 0.706 | 5.68×10−3 | 4.38×10−2 | 0.368 | 0.226 | 0.766 | 4.65×10−4 |
| cg22619018 | CSMD1 | 8.17×10−3 | 0.433 | 0.420 | 0.854 | 1.66×10−3 | 3.14×10−2 | 0.545 | 0.211 | 0.989 | 2.40×10−4 |
| cg12629325 | PCDHAC1 | 3.28×10−2 | 0.409 | 0.442 | 0.758 | 6.56×10−3 | 4.47×10−2 | 0.539 | 0.349 | 0.804 | 5.81×10−4 |
| cg17872757 | FLI1 | 6.86×10−2 | 0.582 | 0.643 | 0.761 | 5.02×10−3 | 2.94×10−2 | 0.680 | 0.443 | 0.794 | 4.83×10−4 |
| cg13562911 | ELOVL2-AS1 | 8.84×10−3 | 0.346 | 0.205 | 0.742 | 4.64×10−3 | 3.73×10−2 | 0.433 | 0.151 | 0.729 | 7.11×10−4 |
| cg18671950 | FBN1 | 3.70×10−2 | 0.357 | 0.635 | 0.955 | 2.89×10−3 | 8.95×10−2 | 0.464 | 0.326 | 0.883 | 4.31×10−4 |
| cg25583174 | FGF2 | 2.17×10−2 | 0.311 | 0.279 | 0.913 | 2.77×10−3 | 6.89×10−2 | 0.426 | 0.230 | 0.857 | 3.44×10−4 |
| cg20415809 | ITGA4 | 4.36×10−2 | 0.365 | 0.521 | 0.803 | 3.28×10−3 | 1.39×10−2 | 0.476 | 0.365 | 0.810 | 3.59×10−4 |
| cg16778809 | ADAM23 | 8.41×10−3 | 0.867 | 0.717 | 0.916 | 2.81×10−3 | 5.10×10−2 | 0.993 | 0.500 | 0.824 | 4.64×10−4 |
| Probe IDa | Gene symbol | Recurrenceb | Disease-related deathb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | ||
| cg24687051 | KCNQ5 | 4.75×10−2 | 0.587 | 0.647 | 0.846 | 4.52×10−3 | 0.177 | 0.613 | 0.384 | 0.815 | 6.25×10−4 |
| cg03168582 | DMRT1 | 1.50×10−4 | 0.774 | 0.612 | 0.961 | 1.71×10−3 | 6.05×10−3 | 0.841 | 0.356 | 0.910 | 3.33×10−4 |
| cg07080358 | CNRIP1 | 3.64×10−2 | 0.444 | 0.670 | 0.842 | 2.29×10−3 | 7.49×10−2 | 0.501 | 0.458 | 0.816 | 3.41×10−4 |
| cg11939071 | DTX1 | 2.37×10−3 | 0.672 | 0.141 | 0.824 | 2.18×10−3 | 5.37×10−2 | 0.662 | 0.126 | 0.941 | 4.25×10−4 |
| cg22029275 | AMER2 | 5.30×10−2 | 0.270 | 0.328 | 0.706 | 5.68×10−3 | 4.38×10−2 | 0.368 | 0.226 | 0.766 | 4.65×10−4 |
| cg22619018 | CSMD1 | 8.17×10−3 | 0.433 | 0.420 | 0.854 | 1.66×10−3 | 3.14×10−2 | 0.545 | 0.211 | 0.989 | 2.40×10−4 |
| cg12629325 | PCDHAC1 | 3.28×10−2 | 0.409 | 0.442 | 0.758 | 6.56×10−3 | 4.47×10−2 | 0.539 | 0.349 | 0.804 | 5.81×10−4 |
| cg17872757 | FLI1 | 6.86×10−2 | 0.582 | 0.643 | 0.761 | 5.02×10−3 | 2.94×10−2 | 0.680 | 0.443 | 0.794 | 4.83×10−4 |
| cg13562911 | ELOVL2-AS1 | 8.84×10−3 | 0.346 | 0.205 | 0.742 | 4.64×10−3 | 3.73×10−2 | 0.433 | 0.151 | 0.729 | 7.11×10−4 |
| cg18671950 | FBN1 | 3.70×10−2 | 0.357 | 0.635 | 0.955 | 2.89×10−3 | 8.95×10−2 | 0.464 | 0.326 | 0.883 | 4.31×10−4 |
| cg25583174 | FGF2 | 2.17×10−2 | 0.311 | 0.279 | 0.913 | 2.77×10−3 | 6.89×10−2 | 0.426 | 0.230 | 0.857 | 3.44×10−4 |
| cg20415809 | ITGA4 | 4.36×10−2 | 0.365 | 0.521 | 0.803 | 3.28×10−3 | 1.39×10−2 | 0.476 | 0.365 | 0.810 | 3.59×10−4 |
| cg16778809 | ADAM23 | 8.41×10−3 | 0.867 | 0.717 | 0.916 | 2.81×10−3 | 5.10×10−2 | 0.993 | 0.500 | 0.824 | 4.64×10−4 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bP values of <0.05 are underlined.
cAverage DNA methylation levels in T samples (cut-off value ≤βT or >βT).
dHistological classification (differentiated, undifferentiated or mucin-producing). If the tumor showed heterogeneity, findings in the predominant area were described.
eHistological classification (differentiated, undifferentiated or mucin-producing). If the tumor showed heterogeneity, the most aggressive features of the tumor were described.
fTumor stage (pT1–pT2 or pT3–pT4).
gPathological Tumor-Node-Metastasis stage (IA–IB, IIA–IIB or IIIA–IV).
Multivariate analyses using the Cox proportional hazards regression model for recurrence and disease-related death of patients with gastric cancers
| Probe IDa | Gene symbol | Recurrenceb | Disease-related deathb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | ||
| cg24687051 | KCNQ5 | 4.75×10−2 | 0.587 | 0.647 | 0.846 | 4.52×10−3 | 0.177 | 0.613 | 0.384 | 0.815 | 6.25×10−4 |
| cg03168582 | DMRT1 | 1.50×10−4 | 0.774 | 0.612 | 0.961 | 1.71×10−3 | 6.05×10−3 | 0.841 | 0.356 | 0.910 | 3.33×10−4 |
| cg07080358 | CNRIP1 | 3.64×10−2 | 0.444 | 0.670 | 0.842 | 2.29×10−3 | 7.49×10−2 | 0.501 | 0.458 | 0.816 | 3.41×10−4 |
| cg11939071 | DTX1 | 2.37×10−3 | 0.672 | 0.141 | 0.824 | 2.18×10−3 | 5.37×10−2 | 0.662 | 0.126 | 0.941 | 4.25×10−4 |
| cg22029275 | AMER2 | 5.30×10−2 | 0.270 | 0.328 | 0.706 | 5.68×10−3 | 4.38×10−2 | 0.368 | 0.226 | 0.766 | 4.65×10−4 |
| cg22619018 | CSMD1 | 8.17×10−3 | 0.433 | 0.420 | 0.854 | 1.66×10−3 | 3.14×10−2 | 0.545 | 0.211 | 0.989 | 2.40×10−4 |
| cg12629325 | PCDHAC1 | 3.28×10−2 | 0.409 | 0.442 | 0.758 | 6.56×10−3 | 4.47×10−2 | 0.539 | 0.349 | 0.804 | 5.81×10−4 |
| cg17872757 | FLI1 | 6.86×10−2 | 0.582 | 0.643 | 0.761 | 5.02×10−3 | 2.94×10−2 | 0.680 | 0.443 | 0.794 | 4.83×10−4 |
| cg13562911 | ELOVL2-AS1 | 8.84×10−3 | 0.346 | 0.205 | 0.742 | 4.64×10−3 | 3.73×10−2 | 0.433 | 0.151 | 0.729 | 7.11×10−4 |
| cg18671950 | FBN1 | 3.70×10−2 | 0.357 | 0.635 | 0.955 | 2.89×10−3 | 8.95×10−2 | 0.464 | 0.326 | 0.883 | 4.31×10−4 |
| cg25583174 | FGF2 | 2.17×10−2 | 0.311 | 0.279 | 0.913 | 2.77×10−3 | 6.89×10−2 | 0.426 | 0.230 | 0.857 | 3.44×10−4 |
| cg20415809 | ITGA4 | 4.36×10−2 | 0.365 | 0.521 | 0.803 | 3.28×10−3 | 1.39×10−2 | 0.476 | 0.365 | 0.810 | 3.59×10−4 |
| cg16778809 | ADAM23 | 8.41×10−3 | 0.867 | 0.717 | 0.916 | 2.81×10−3 | 5.10×10−2 | 0.993 | 0.500 | 0.824 | 4.64×10−4 |
| Probe IDa | Gene symbol | Recurrenceb | Disease-related deathb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | β values in T samplesc | Predominant histologyd | Most aggressive histologye | pTf | TNM stageg | ||
| cg24687051 | KCNQ5 | 4.75×10−2 | 0.587 | 0.647 | 0.846 | 4.52×10−3 | 0.177 | 0.613 | 0.384 | 0.815 | 6.25×10−4 |
| cg03168582 | DMRT1 | 1.50×10−4 | 0.774 | 0.612 | 0.961 | 1.71×10−3 | 6.05×10−3 | 0.841 | 0.356 | 0.910 | 3.33×10−4 |
| cg07080358 | CNRIP1 | 3.64×10−2 | 0.444 | 0.670 | 0.842 | 2.29×10−3 | 7.49×10−2 | 0.501 | 0.458 | 0.816 | 3.41×10−4 |
| cg11939071 | DTX1 | 2.37×10−3 | 0.672 | 0.141 | 0.824 | 2.18×10−3 | 5.37×10−2 | 0.662 | 0.126 | 0.941 | 4.25×10−4 |
| cg22029275 | AMER2 | 5.30×10−2 | 0.270 | 0.328 | 0.706 | 5.68×10−3 | 4.38×10−2 | 0.368 | 0.226 | 0.766 | 4.65×10−4 |
| cg22619018 | CSMD1 | 8.17×10−3 | 0.433 | 0.420 | 0.854 | 1.66×10−3 | 3.14×10−2 | 0.545 | 0.211 | 0.989 | 2.40×10−4 |
| cg12629325 | PCDHAC1 | 3.28×10−2 | 0.409 | 0.442 | 0.758 | 6.56×10−3 | 4.47×10−2 | 0.539 | 0.349 | 0.804 | 5.81×10−4 |
| cg17872757 | FLI1 | 6.86×10−2 | 0.582 | 0.643 | 0.761 | 5.02×10−3 | 2.94×10−2 | 0.680 | 0.443 | 0.794 | 4.83×10−4 |
| cg13562911 | ELOVL2-AS1 | 8.84×10−3 | 0.346 | 0.205 | 0.742 | 4.64×10−3 | 3.73×10−2 | 0.433 | 0.151 | 0.729 | 7.11×10−4 |
| cg18671950 | FBN1 | 3.70×10−2 | 0.357 | 0.635 | 0.955 | 2.89×10−3 | 8.95×10−2 | 0.464 | 0.326 | 0.883 | 4.31×10−4 |
| cg25583174 | FGF2 | 2.17×10−2 | 0.311 | 0.279 | 0.913 | 2.77×10−3 | 6.89×10−2 | 0.426 | 0.230 | 0.857 | 3.44×10−4 |
| cg20415809 | ITGA4 | 4.36×10−2 | 0.365 | 0.521 | 0.803 | 3.28×10−3 | 1.39×10−2 | 0.476 | 0.365 | 0.810 | 3.59×10−4 |
| cg16778809 | ADAM23 | 8.41×10−3 | 0.867 | 0.717 | 0.916 | 2.81×10−3 | 5.10×10−2 | 0.993 | 0.500 | 0.824 | 4.64×10−4 |
aProbe ID for the Infinium HumanMethylation27 Bead Array.
bP values of <0.05 are underlined.
cAverage DNA methylation levels in T samples (cut-off value ≤βT or >βT).
dHistological classification (differentiated, undifferentiated or mucin-producing). If the tumor showed heterogeneity, findings in the predominant area were described.
eHistological classification (differentiated, undifferentiated or mucin-producing). If the tumor showed heterogeneity, the most aggressive features of the tumor were described.
fTumor stage (pT1–pT2 or pT3–pT4).
gPathological Tumor-Node-Metastasis stage (IA–IB, IIA–IIB or IIIA–IV).
Correlation between DNA methylation and mRNA expression
The DNA methylation levels of the OLFM4 (r = −0.6221 and P = 6.90×10−4), KCNQ5 (r = −0.5243 and P = 5.00×10−3), FBN1 (r = −0.3339, P = 2.18×10−2) and ITGA4 (r = −0.5192 and P = 9.69×10−3) genes revealed by the Infinium assay were inversely correlated with the levels of mRNA expression revealed by real-time quantitative RT-PCR in T and N samples (Supplementary Figure 3, available at Carcinogenesis Online). In addition, the ADAM23 (r = −0.3027) gene also tended to show an inverse correlation between the level of DNA methylation and that of mRNA expression, although this tendency did not reach a statistically significant level (Supplementary Figure 3, available at Carcinogenesis Online).
Discussion
Here we have reported the results of the Infinium assay for 214 samples of gastric tissue (109 N and 105 T samples). As the field cancerization concept has been accepted in the context of the stomach, N samples obtained from patients with gastric carcinomas may be at the precancerous stage. Therefore, we focused on DNA methylation status at the precancerous stage (βN). Based on βN data for 3661 probes (Supplementary Table 4, available at Carcinogenesis Online) associated with gastric carcinogenesis, epigenetic clustering of gastric carcinomas was observed (Figure 1A). Even though such clustering was established on the basis of DNA methylation profiles at the precancerous stage, it was significantly correlated with the clinicopathological aggressiveness (in terms of an undifferentiated histological type, deeper invasion and/or higher pathological TNM stage [Table 1]) of established tumors. Moreover, the epigenetic clustering based on βN was significantly correlated with patient outcome (Figure 1B). In this study, this impact on patient outcome was strictly confirmed by long-term follow-up (Figure 1B). These data indicated that distinct DNA methylation profiles, which may determine tumor aggressiveness and patient outcome, have already become established at the precancerous stage. These findings are compatible with those of our previous studies of the kidney (22,23), lung (16,17), urinary bladder (24), liver (25) and pancreas (26), for which DNA methylation profiles determining tumor aggressiveness and patient outcome have already been established in non-cancerous tissues at the precancerous stage.
Although the incidence of H. pylori infection in Cluster B1 (70%) tended to be higher than in Cluster A1 (55%), no statistically significant correlation was evident between H.pylori infection and epigenetic clustering (Table 1). Although patient age was significantly correlated with epigenetic clustering (Table 1), no significant correlation between patient age and H.pylori infection was observed in the present cohort (Supplementary Table 7, available at Carcinogenesis Online). However, patient age was significantly correlated with intestinal metaplasia in the non-cancerous gastric mucosa, reflecting the long history of H.pylori infection, subsequent chronic active gastritis and atrophic gastritis (33) (Supplementary Table 7, available at Carcinogenesis Online). On the other hand, genes previously reported to show age-related methylation, such as GDNF, CDH1, RARB2, CDH13, MYOD1, SFRP1, SLC16A12, DPYS and TUSC3 (34), have not been listed as hallmark genes characterizing epigenetic clustering in Table 2. Taken together, the data suggest that any significant correlation between patient age and epigenetic clustering may not depend solely on H.pylori infection or age-related methylation of specific genes. DNA methylation profiles that determine tumor aggressiveness and patient outcome may become established through long-term accumulation of effects resulting from H.pylori infection, subsequent chronic active gastritis, atrophic gastritis and intestinal metaplasia.
After identification of the hallmark genes in N samples characterizing the epigenetic clustering, we examined whether DNA methylation profiles in those samples were inherited by the gastric carcinomas themselves. We then focused on DNA methylation levels of 60 hallmark genes in T samples selected on the basis of βN values (Table 2), and again found that these DNA methylation levels were significantly correlated with the clinicopathological aggressiveness (undifferentiated histological type, deeper invasion and/or higher pathological TNM stage [Table 3]) of the tumors and patient outcome (Figure 2 and Supplementary Table 4, available at Carcinogenesis Online), reflecting the correlations observed for methylation profiles at the precancerous stages (Figure 1B; Table 1). Moreover, the DNA methylation levels of the hallmark genes in T samples were prognostically independent of clinicopathological aggressiveness. Among the 60 hallmark genes selected on the basis of βN, 23 genes included in Table 2A and 25 other genes included in Table 2B (48 genes in total) were included in Tables 3 and 4, Supplementary Table 4 (available at Carcinogenesis Online) and/or Figure 2: thus, the DNA methylation levels of most of the 60 hallmark genes in T samples actually had clinicopathological and prognostic impact. These data indicated that DNA methylation profiles at the precancerous stages determining tumor aggressiveness and patient outcome were inherited by the gastric carcinomas themselves.
It is feasible that a number of genes previously reported to be methylated in human cancers were included in the above 48 hallmark genes whose DNA methylation status had clinicopathological and prognostic impact. For example, we have reported that the PCDHAC1 gene, included in Tables 4, Supplementary Table 6 (available at Carcinogenesis Online) and Figure 2, is one of the CIMP (CpG island methylator phenotype) marker genes in renal cell carcinomas (22). DNA methylation of the CSMD1 and FBN1 genes, again included in Tables 3 and 4, Supplementary Table 6 (available at Carcinogenesis Online) and/or Figure 2, has been reported in human colorectal cancers (35,36), head and neck cancers (37) and malignant lymphomas (38). DNA methylation of the KCNQ5 (39), FLI1 (40), ITGA4 (41) and ADAM23 (42) genes, which appeared in Table 4, Supplementary Table 6 (available at Carcinogenesis Online) and/or Figure 2, has also been reported in human stomach cancers and cancers derived from other organs.
On the other hand, with regard to the ELOVL2-AS1, SLC3A1, LOC389641 and BEND5 genes included in Tables 3 and 4, Supplementary Table 6 (available at Carcinogenesis Online) and/or Figure 2 , no functional implication in carcinogenesis has yet been revealed, and no DNA methylation alterations have been reported in human cancers. Therefore, the functions and regulatory mechanisms of these genes should be further examined in relation to gastric carcinogenesis.
A number of tumor-related genes were also included among the hallmark genes whose βT values had clinicopathological and prognostic impact, and are listed in Tables 3 and 4, Supplementary Table 6 (available at Carcinogenesis Online) and/or Figure 2. For example, with regard to the above-mentioned ADAM23 gene, its metalloprotease domain is inactive. Instead, it has been reported that ADAM23 specifically interacts with αvβ3 integrin via its disintegrin domain and negatively regulates the metastasis-promoting potential of αvβ3 integrin during cancer progression (43). OLFM4 binds to the potent apoptosis inducer GRIM-19 and promotes proliferation of cancer cells by favoring transition from the S to the G2/M phase (44). The adenomatous polyposis coli membrane recruitment (Amer) family protein, AMER2, is one of binding partners of the adenomatous polyposis coli tumor suppressor protein, and acts as a negative regulator in the Wnt/β-catenin signaling cascade (45). The GPSM1 gene encodes a member of the activator of G-protein signaling protein family. In multiple myeloma cells, GPSM1 has been shown to exert anti-apoptosis activity by enhancing phosphorylation of the cyclic AMP response element-binding protein, CREB (46). The CCL28 gene encodes a chemokine ligand. Tumor hypoxia promotes the recruitment of regulatory T cells through induction of CCL28 expression, resulting in immune tolerance and tumor angiogenesis (47). The DTX1 gene encodes a positive regulator of the Notch signaling pathway, and activates mitosis, proliferation and invasion of glioblastoma cells in vitro (48). Moreover, the expression level of DTX1 is reportedly correlated with the outcome of patients with glioblastoma (48). COL23A1 is known to be one of the transmembrane collagens. Expression of the COL23A1 gene is not only a biomarker of non-small cell lung cancer (49) but is also reportedly associated with recurrence and distant metastasis of prostate cancer (50).
There are two possible ways of interpreting the available data: (i) the DNA methylation status of at least a proportion of the above-mentioned tumor-related genes may simply be a surrogate marker of tumor aggressiveness and patient outcome in our gastric cancer cohort, or (ii) DNA methylation of those genes actually participates in the malignant progression of gastric cancer through regulation of expression. Among the genes examined, the DNA methylation levels of OLFM4, KCNQ5, FBN1 and ITGA4 were inversely correlated with their levels of mRNA expression in tissue specimens (Supplementary Figure 3, available at Carcinogenesis Online). The ADAM23 gene also showed such a tendency for inverse correlation (Supplementary Figure 3, available at Carcinogenesis Online), suggesting that DNA methylation may regulate the expression level of such genes. Moreover, knockdown of the OLMF4 gene using small interfering RNA (siRNA) resulted in reduced cell viability of the gastric cancer cell lines NSC-15CF and NSC-4X1a revealed by MTS assay (Supplementary Methods and Supplementary Figure 4, available at Carcinogenesis Online). Knockdown of the ADAM23 gene in the MKN45 gastric cancer cell line resulted in enhanced cell adhesion, which is possibly mediated by integrins and frequently involved in cancer invasion and metastasis (Supplementary Methods and Supplementary Figure 4, available at Carcinogenesis Online). These findings support the above possibility (ii), i.e. that DNA methylation of specific genes actually participates in the malignant progression of gastric cancer through regulation of gene expression.
In the case of either (i) or (ii), even at the precancerous stage, the DNA methylation profiles of such tumor-related genes already show characteristic epigenetic clustering, reflecting differences in prognosis among patients. Therefore, accumulated effects resulting from H.pylori infection, subsequent chronic active gastritis, atrophic gastritis, intestinal metaplasia and other carcinogenetic factors induce distinct DNA methylation profiles during the process of field cancerization, and such profiles at the precancerous stage are inherited by the gastric cancers themselves, thus determining tumor aggressiveness and patient outcome.
Supplementary material
Supplementary Table 1–7 and Supplementary Data can be found at Supplementary Data
Funding
National Institute of Biomedical Innovation (NiBio) , 10-41, 10-42; Japan Society for the Promotion of Science (JSPS) (23390096, 25460487); National Cancer Center Research and Development Fund (26-A-1); the Applied Research for Innovative Treatment of Cancer (H26-019).
Conflict of Interest Statement: None declared.
Abbreviations
- EB virus
Epstein–Barr virus
- H. pylori
Helicobacter pylori
- N
non-cancerous mucosa
- ROC
receiver operating characteristic
- T
tumorous tissue
- TNM
tumor node metastasis
References

{kind=link}
{kind=link}