




In their recent article in Brain, Ravenscroft and Cabrera-Serrano1 discuss the expanding genetic spectrum of hyperCKaemia. As explained by the authors, there are numerous cellular processes, which may be affected in these disorders, leading to release of creatine kinase (CK) into the circulation. One potential reason for chronic hyperCKaemia is caveolar dysfunction. Caveolae are flask-shaped invaginations of the sarcolemmal membrane, which constitute microdomains for specific signalling molecules. Moreover, they provide a membrane reservoir that is critical for the necessary increase in surface area during stretch of muscle fibres.2,3 Thereby, caveolae preserve muscle cell integrity in the event of membrane tension and mechanical stress.
Disorders of caveolar function and integrity are mainly caused by pathogenic variants in the CAV3 gene, which cause a continuum of muscle diseases named ‘caveolinopathies’ (rippling muscle disease-2, OMIM 606072; Tateyama type of distal myopathy, OMIM 614321; hyperCKaemia syndrome, OMIM 123320).
Clinically, caveolinopathies are characterized by slowly progressive muscle weakness, exercise intolerance and elevated CK levels. Moreover, hypertrophic skeletal muscles and generalized rapid muscle contractions may occur in some individuals.
Experimental studies in cell culture systems and knockout mice have suggested that the protein PACSIN3 (protein kinase C and casein kinase substrate in neurons 3; also known as syndapin III) is also involved in caveolar biogenesis.3 Studies in heart muscles of Pacsin3 knockout mice revealed a reduction of deeply invaginated caveolae with a concomitant decrease of caveolin-3 density in these regions. Physical exercise in Pacsin3 knockout mice resulted in the presence of hypertrophic and necrotic muscle fibres.3
Here, we report on three individuals from two unrelated families presenting with delayed motor development, exercise intolerance and hyperCKaemia. Exome sequencing identified biallelic loss-of-function variants in PACSIN3 as the underlying cause.
Materials and methods
Ethics approval
The institutional review boards of the Heinrich Heine University Düsseldorf approved this study (#2021–1340). Participants gave informed consent to participate in the study before taking part.
Cell culture
Fibroblasts were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (all from Life Technologies) at 37°C in a humidified atmosphere of 5% CO2.
Immunoblotting
Fibroblasts were detached with TrypLE™ Select and lysed using CelLytic™ M reagent with protease inhibitors. Protein lysates were separated on 4–12% Bis/Tris gels (Invitrogen) with MOPS running buffer. For each lane, 30 µg of protein was loaded. Blotting was performed on nitrocellulose membranes. Blocking was done with 3% milk powder in TBS with 0.1% Tween. The following primary antibodies were used: rabbit anti PACSIN3 (#10639-1-AP, Proteintech, 1:1000) and mouse anti GAPDH (AM4300, Invitrogen, 1:4000). Incubation was carried out overnight at 4°C. As secondary antibodies, anti-rabbit IgG HRP (Amersham NA 934 V) and anti-mouse IgG HRP (Amersham NA 931 V) were used for 1 h at room temperature. BM Chemiluminescence blotting substrate (Roche) was used for visualization. Results were recorded using Chemidoc (Bio-Rad). Bands were quantified using Image Lab software V 5.2.1 (Bio-Rad). Statistical significance was assessed using t-test.
Caveolin-3 immunostaining
Caveolin-3 immunostaining was carried out as previously described.4
Electron microscopy
Muscle tissue was fixed in 6% buffered glutaraldehyde and embedded. Ultrathin sections (100 nm) were mounted on grids and examined by transmission electron microscopy.5
Genetic investigations
Family 1
Exome sequencing (ES) was conducted on genomic DNA from peripheral blood leucocytes, as previously described.6 In brief, coding genomic regions were enriched using a SureSelect XT Human All Exon Kit V7 (Agilent Technologies) and sequenced as 2 × 115 bp paired-end reads on a NovaSeq6000 system (Illumina) to an average 198× coverage of the target region. Generated sequences were analysed using the megSAP analysis pipeline (https://github.com/imgag/megSAP).
Family 2
Individual 2A underwent chromosomal microarray analysis using the Affymetrix Cytoscan XON Assay kit (ThermoFisher Scientific) and clinical exome sequencing analysis using the TruSight One Expanded Sequencing Panel kit (Illumina Inc.). Subsequently, research-based exome sequencing was performed on Individual 2A's peripheral blood sample using the Twist Exome 2.0 kit (Twist Bioscience) on the NovaSeq 6000 platform.
Results
Case reports
Family 1
Individual 1 (female) was the first child of healthy, non-consanguineous parents with German ancestry (pedigree in Fig. 1A). She was born preterm at 32 gestational weeks. During the first months of life, she had problems with constipation, feeding difficulties and poor weight gain. Muscular hypotonia became evident. Achievement of motor milestones was moderately delayed (free walking with 2 years). Parents reported early fatigue, muscle pain and exercise intolerance. Cognitive development was normal. Examination at the age of 5 years revealed normal muscle tone and strength. No muscle rippling, muscle mounding or PIRCS (percussion induced repetitive contractions) were observed. Episodes of fatigue, muscle pain and malaise were variable. Laboratory work-up revealed elevated CK with values between 1000 and 1800 U/l (controls: <228) and CK-MB between 80 and 90 U/l (controls: <25). Transaminases were mildly elevated (GOT 72 U/l, controls: <52, GPT 51 U/l, controls: <29). Investigation of CK levels in the parents was normal. Cardiac work-up was normal. Muscle ultrasound was without pathological findings. Metabolic investigations, including screening for disorders of fatty acid oxidation and Pompe disease, detected no abnormalities. Muscle biopsy was performed at the age of 5 years. Examination revealed variation in muscle fibre diameter but no specific abnormalities.
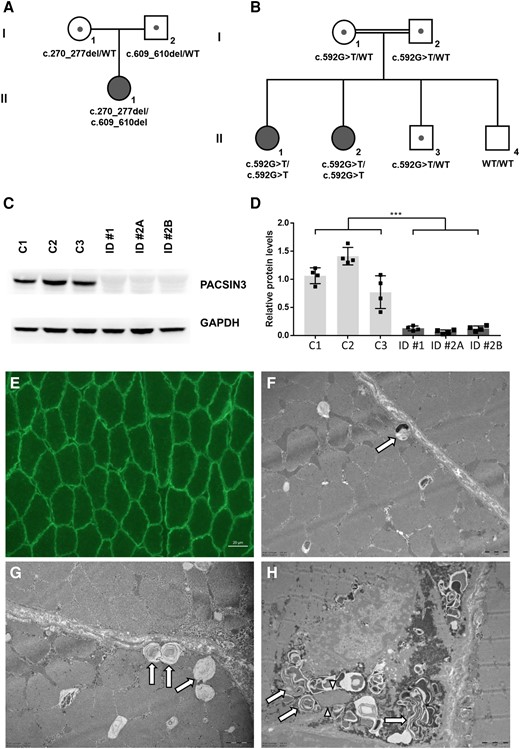
Genetic, biochemical and histological findings in individuals with PACSIN3 deficiency. (A and B) Pedigrees of Family 1 (A) and Family 2 (B) with pathogenic variants in PACSIN3. Mutation status of affected (filled symbols) and healthy (open symbols) family members is shown. (C) Representative immunoblotting image of patient-derived (ID 1, ID 2A, ID 2B) and control (C1, C2, C3; age-matched, female) fibroblasts. In patient-derived cell lines, PACSIN3 protein is not detectable. The corresponding unmodified full-length blots can be found in the Supplementary material. (D) Quantitated density of immunoblot bands from four independent experiments. Average values are presented as the mean ± standard error of the mean (SEM); ***P < 0.001 significantly different from controls. (E) Immunofluorescence of caveolin-3 in biopsied muscle tissue derived from Individual 1, showing normal pattern of sarcolemmal immunoreactivity (scale bar = 20 µm). (F–H) Electron microscopy images of biopsied muscle fibres from Individual 1. (F and G) Abnormal vacuoles preferentially found in close proximity to the sarcolemma (arrows), containing membranous material, which in some of the vacuoles coalesces to form membranous cytoplasmic bodies (scale bars = 1 µm). (H) Focal subsarcolemmal build-up of glycogen granules and of membranous cytoplasmic bodies combined with proliferations of membranous tubules forming loops (arrows) and tubular aggregate-like stacks (arrowheads) (scale bar = 2 µm). WT = wild-type.
Family 2
Individuals 2A and 2B (both females) were born at term after uneventful pregnancies to healthy, first-degree consanguineous parents with Turkish ancestry (Fig. 1B). Two siblings were healthy. Developmental milestones were achieved in time, but parents noticed exercise intolerance and early fatigue. Neurological examinations of both children were unremarkable, with normal muscle strength, muscle tone and reflexes. No muscle mounding or PIRCS were observed. Individual 2A had elevated CK levels ranging from 649 to 1291 IU/l (controls: 44–189) and normal transaminase levels. Individual 2B also had elevated CK levels ranging from 769 to 991 IU/l. Levels of GPT and GOT were slightly elevated (values of 55 and 56 IU/l, controls: <36). Nerve conduction studies and EMG of both patients were normal. Respiratory function tests, echocardiography and ECG examinations were without pathological findings. Histopathological examination of muscle tissue derived from Individual 2A at the age of 9 years showed a mild variation in fibre diameter, few atrophic fibres and internal nuclei in some fibres. Muscle biopsy examination of Individual 2B at the age of 7 years revealed similar findings with more frequent internal nuclei and focal increase in endomysial connective tissue. No additional anomalies were detected in the histological and the immunohistochemical evaluations. Follow-up examinations at the current age of 11.5 and 9.5 years revealed no additional clinical features.
Genetic findings
Family 1
Genetic testing for dystrophinopathies was normal. Exome sequencing with stringent filtering for rare (minor allele frequency 1% in in-house and public databases), high impact variants prioritized two heterozygous nonsense variants in PACSIN3 [ENST00000298838.11, NM_016223.5:c.270_277del, p.(Leu91AlafsTer15) and c.609_610del, p.(Lys203AsnfsTer4)]. Both variants are rare (three and 30 heterozygous calls in the database gnomAD v4.0.0, respectively) and absent from an in-house database containing >22 000 exome or genome datasets. Of note, no other predicted loss-of-function PACSIN3 variants are listed in a homozygous state in these databases. Sanger sequencing confirmed a compound heterozygous state of the variants with the parents being heterozygous carriers. According to the positions of the nonsense variants (exons 5 and 7, respectively, with a total of 11 exons), both aberrant mRNA species are prone to nonsense-mediated decay.
Family 2
Genetic investigations focusing on aetiology revealed that both patients had normal results in Duchenne muscular dystrophy (DMD) multiplex ligation-dependent probe amplification (MLPA) analysis, chromosomal microarray analysis and clinical exome sequencing. Upon research-based exome sequencing analysis of Individual 2A's peripheral blood sample, a stop codon variant in the PACSIN3 gene, NM_016223.5:c.592G>T, p.(Glu198Ter), was identified in a homozygous state. Individual 2B was also found to be homozygous for the PACSIN3 variant by Sanger sequencing. Additionally, the parents and Individual 2C in the family were heterozygous for the PACSIN3 variant, while Individual 2D was found to be wild-type.
PACSIN3 immunoblotting
Immunoblotting revealed absence of PACSIN3 in patient-derived fibroblasts compared to age- and sex-matched healthy controls (Fig. 1C and D).
Caveolin-3 immunostaining
No abnormalities in caveolin-3 immunoreactivity were observed in the muscle tissue of Individual 1 (Fig. 1E).
Electron microscopy
Ultrastructural studies of muscle tissue from Individual 1 revealed subsarcolemmal build-up of vacuoles containing membranous electron-dense material. Occasionally, these vacuoles transit into membranous cytoplasmic bodies indicative of altered autophagy (Fig. 1F and G). Membranous cytoplasmic bodies were occasionally building up together with focal proliferations of membranous tubules, some of which were forming loops and others were arranged in tubular aggregate-like stacks (Fig. 1H).
Discussion
PACSINs play a role in cytoskeleton organization and intracellular trafficking.7 The PACSIN protein family comprises three members: PACSIN1, PACSIN2 and PACSIN3. PACSIN1 is almost exclusively expressed in the brain, while PACSIN2 is ubiquitously expressed. PACSIN3 is mainly expressed in skeletal muscle, heart and lung.7 To date, there is limited knowledge about the implication of PACSINs in human diseases.
The biological function of PACSIN3 was studied in mice, showing that Pacsin3 knockout impaired formation of deep membrane invaginations and was associated with a lower density of caveolin-3 in the deeply invaginated caveolae in heart muscle.3 In skeletal muscle, mechanical stress resulted in muscle damage, which was reminiscent of human muscle pathology caused by pathogenic variants in the CAV3 gene.3,7
Our study now complements these data and suggests that PACSIN3-deficiency extends the spectrum of human caveolinopathies. The clinical phenotype and laboratory findings of the individuals reported here are well in line with the clinical spectrum of caveolinopathies. Genetic and immunoblotting data indicate a loss-of-function effect as the underlying cause. The fact that immunofluorescence-based analysis of caveolin-3 abundance in muscle tissue of PACSIN3 patients was normal does not argue against the pathogenicity of the PACSIN3 variants. Studies in heart muscle of Pacsin3 knockout mice also revealed that the overall staining of caveolin-3 at the plasma membrane was not altered.3 One might speculate that PACSIN3-related control of caveolae-maintenance occurs independent of caveolin-3 via interaction with other proteins localized within these microdomains.
Ultrastructural studies in muscle tissue derived from Individual 1 showed accumulation of membranous tubules, some of which were arranged in tubular aggregates. It is known that caveolin-3 defects are associated with impaired cellular transport processes leading to protein mislocalization with Golgi retention and endoplasmic reticulum stress.8 A consequence may be the build-up of vacuoles indicative for altered autophagy, as revealed by our studies. This pathomorphological finding is in line with the known function of different caveolae-associated proteins in the modulation of autophagy and the findings obtained in in vitro and in vivo systems of CAV3-related caveolinopathy.8-10
At the current stage, the exact mechanism by which PACSIN3 deficiency leads to impaired caveolar biogenesis is still unresolved. Mechanistically, caveolin-3 and PACSIN3 play a role in the same pathways (e.g. intracellular trafficking, protein processing), suggesting a functional overlap.
In summary, our results indicate the importance of PACSIN3 for the structural integrity of muscle fibres in humans and show that recessive loss-of-function variants may cause a myopathy with hyperCKaemia in children.
Data availability
The main data associated with this study are present in the article or Supplementary material. Additional raw data are available from the corresponding author, upon reasonable request.
Acknowledgements
We thank the patients and their families for participating in this study. This work was partly done within the Zentrum für Seltene Erkrankungen of the University Hospital Düsseldorf (ZSED). F.D. is a member of the European Reference Network for Rare Hereditary Metabolic Disorders MetabERN. We would like to thank Prof. Esra Tuğ and the cytogenetics team of the Department of Medical Genetics, Faculty of Medicine, Gazi University, for their assistance in cell culture.
Funding
The study was supported by a grant of the German Research Foundation/Deutsche Forschungsgemeinschaft (DI 1731/2-3 to F.D.) and the ‘Elterninitiative Kinderkrebsklinik e.V.’ (Düsseldorf; #701900167). A.R. and U.S.S. appreciate the financial support of the Deutsche Gesellschaft für Muskelkranke (DGM).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
References
Author notes
These authors contributed equally to this work.


{kind=link}