




Abstract
Vascular dysfunction is increasingly recognized as an important contributor to the pathogenesis of Alzheimer’s disease. Alterations in vascular endothelial growth factor (VEGF) pathways have been implicated as potential mechanisms. However, the specific impact of VEGF proteins in preclinical Alzheimer’s disease and their relationships with other Alzheimer’s disease and vascular pathologies during this critical early period remain to be elucidated.
We included 317 older adults from the Harvard Aging Brain Study, a cohort of individuals who were cognitively unimpaired at baseline and followed longitudinally for up to 12 years. Baseline VEGF family protein levels (VEGFA, VEGFC, VEGFD, PGF and FLT1) were measured in fasting plasma using high-sensitivity immunoassays. Using linear mixed effects models, we examined the interactive effects of baseline plasma VEGF proteins and amyloid PET burden (Pittsburgh Compound-B) on longitudinal cognition (Preclinical Alzheimer Cognitive Composite-5). We further investigated if effects on cognition were mediated by early neocortical tau accumulation (flortaucipir PET burden in the inferior temporal cortex) or hippocampal atrophy. Lastly, we examined the impact of adjusting for baseline cardiovascular risk score or white matter hyperintensity volume.
Baseline plasma VEGFA and PGF each showed a significant interaction with amyloid burden on prospective cognitive decline. Specifically, low VEGFA and high PGF were associated with greater cognitive decline in individuals with elevated amyloid, i.e. those on the Alzheimer’s disease continuum. Concordantly, low VEGFA and high PGF were associated with accelerated longitudinal tau accumulation in those with elevated amyloid. Moderated mediation analyses confirmed that accelerated tau accumulation fully mediated the effects of low VEGFA and partially mediated (31%) the effects of high PGF on faster amyloid-related cognitive decline. The effects of VEGFA and PGF on tau and cognition remained significant after adjusting for cardiovascular risk score or white matter hyperintensity volume. There were concordant but non-significant associations with longitudinal hippocampal atrophy.
Together, our findings implicate low VEGFA and high PGF in accelerating early neocortical tau pathology and cognitive decline in preclinical Alzheimer’s disease. Additionally, our results underscore the potential of these minimally-invasive plasma biomarkers to inform the risk of Alzheimer’s disease progression in the preclinical population. Importantly, VEGFA and PGF appear to capture distinct effects from vascular risks and cerebrovascular injury. This highlights their potential as new therapeutic targets, in combination with anti-amyloid and traditional vascular risk reduction therapies, to slow the trajectory of preclinical Alzheimer’s disease and delay or prevent the onset of cognitive decline.
Introduction
Vascular dysfunction is increasingly recognized as an important and early partner in the pathogenesis of Alzheimer’s disease (AD).1-6 Growing literature has shed light on the contributions of vascular risk factors and cerebrovascular brain injury on the biological and clinical manifestations of AD.4,7-15 In particular, studies from our group have demonstrated synergistic effects of systemic vascular risks and amyloid-β (Aβ) burden on longitudinal tau accumulation, neurodegeneration and cognitive decline in preclinical AD.14-16 The observation of an early vascular-Aβ synergy highlights vascular dysfunction as a promising therapeutic target, in combination with emerging anti-Aβ therapies, to modify the trajectory of AD progression. However, it remains unclear exactly which vascular-related biological pathways are the most promising targets for intervention and whether current vascular risk and injury measures (e.g. white matter hyperintensities) comprehensively account for vascular impacts on AD pathology and cognitive decline. Therefore, to develop novel, targeted therapies, there remains a critical need to identify and understand the biological pathways mediating vascular contributions to AD.
In this context, vascular endothelial growth factor (VEGF) pathways are of key interest, as they are involved in processes critical for brain and vascular health, including angiogenesis, perfusion and endothelial function.17-20 Alterations in VEGF family gene expression and protein levels in brain, CSF and blood have been demonstrated in AD,21-31 highlighting their potential role in AD pathogenesis. However, studies have reported inconsistent directions of associations, even when examining the same tissue compartment. For example, both increased and decreased levels of VEGFA, the best-studied VEGF ligand, have been demonstrated in CSF and blood of individuals with mild cognitive impairment (MCI) and AD dementia.21-26,28,29,32,33 Similarly, higher VEGFA levels have been associated with both protective and detrimental effects on brain ageing.24-27,29-31,33-37 The inconsistent results may be attributed to technical factors such as limited sensitivity and dynamic range in earlier immunoassays, the type of biofluid analysed (e.g. plasma versus serum; with variable release of VEGF proteins in serum samples due to blood clotting38,39) and limited sample size leading to false positives and negatives. In addition, the clinical heterogeneity of the cohorts examined is another potential contributor to the discordant findings. Evidence suggests that VEGF pathway alterations may vary non-linearly as the disease progresses,25,35,36,40-42 potentially reflecting differences underlying resilience mechanisms, early compensatory response and dysregulated processes during advanced disease. It is therefore critical to isolate and understand the role of VEGF proteins in preclinical AD, a disease stage with relative sparing from irreversible neuronal injury and neurodegeneration, when the potential to meaningfully alter the AD cascade is greatest.43,44 In this context, minimally invasive blood-based biomarkers are of particular interest, as they can be widely applied in preclinical populations to inform the risk of downstream AD pathology and cognitive decline.
The current study is designed to address these critical questions, by leveraging the deeply phenotyped Harvard Aging Brain Study (HABS) cohort,45 a group of older individuals who were cognitively unimpaired (CU) at baseline and followed longitudinally with neuroimaging, biofluids, clinical and cognitive assessments. We quantified VEGF proteins that have been implicated in AD and brain ageing. In addition to VEGFA, we measured PGF and soluble FLT1, which have been associated with worse cognitive and functional outcomes,30,46-48 as well as two related VEGF ligands which are relatively understudied in the context of AD and brain ageing (VEGFC and VEGFD). We examined whether these VEGF proteins in baseline plasma have interactive or additive effects with baseline Aβ burden on prospective cognitive decline. We further assessed whether VEGF effects on cognitive decline were mediated by downstream pathologies in the AD cascade, specifically the accumulation of neocortical tau and hippocampal atrophy. Lastly, we examined if VEGF effects are distinct from the impact of systemic vascular risk or cerebral vascular injury and may therefore represent novel targets for intervention.
Materials and methods
Participants
The current study included 317 older CU adults from HABS,45 a cohort study of individuals aged 50 to 90 recruited from the community who were cognitively unimpaired at baseline and assessed longitudinally with neuroimaging, clinical assessments and cognitive testing. We included all participants who had baseline plasma sampling, Aβ PET imaging and at least two longitudinal cognitive assessments. Subsets of participants with longitudinal tau PET (n = 187) and longitudinal MRI (n = 207) were used in PET and MRI analyses, respectively. At study entry, all participants had a global Clinical Dementia Rating49 (CDR) of 0, education-adjusted Mini-Mental State Examination (MMSE)50 score ≥27 and Logical Memory IIa Delayed Recall performance within the normal range.51 Exclusion criteria included a modified Hachinski ischaemic score >4 and a history of stroke with persistent neurological deficits.45 Data were collected from April 2010 through February 2023. The Mass General Brigham Institutional Review Board approved HABS protocol and procedures, and all participants signed a written informed consent prior to the completion of any study procedures.
Plasma VEGF measures
Baseline fasting morning blood samples were collected in BD Vacutainer® EDTA tubes (BD), and plasma was isolated through centrifugation (2000 RCF × 5 min). Aliquoted samples were stored at −80°C until thawed. Plasma samples were analysed on a MESO QuickPlex SQ 120 platform (Meso Scale Diagnostics), a highly sensitive multiplex electrochemiluminescence platform with a broad dynamic range, at the Massachusetts Alzheimer’s Disease Research Center Biomarker Core. We measured five VEGF family proteins (VEGFA, VEGFC, VEGFD, PGF and FLT1) in baseline plasma using the V-PLEX Angiogenesis Panel 1 Human Kit (K15190D, Meso Scale Diagnostics) and used the Angiogenesis Control Pack 1 (C4190, Meso Scale Diagnostics) for plate-to-plate control. Due to our focus on the VEGF pathways, we did not include the remaining two analytes on the angiogenesis kit (FGF and Tie-2) in the current study, as they signal through different mechanisms. It is important to note that the significance of our primary findings would be unaffected if these additional analytes were included in the correction for multiple comparisons. Plasma samples and controls were diluted according to the manufacturer’s recommendations (1:2) and were run in duplicate on each plate. Coefficients of variation (CVs, %) were calculated for sample replicates using the following formula: 100 × (standard deviation/mean). A CV of 20% was used as cut-off criteria for acceptable technical variation, similar to other studies using the same Meso Scale Diagnostics immunoassays.52-54 Plate data were mean normalized using commercial controls to adjust for batch effects, and plate-to-plate adjustment was performed using mean control concentration across all plates as a reference. Normalized concentrations adjusted for dilution factors (range: 0.71–1.20) were reported in pg/ml. All plasma biomarker concentration measures were log10-transformed to account for skewness.
PET imaging
Brain Aβ burden was measured at baseline using 11C-Pittsburgh Compound-B (PiB) PET at the Massachusetts General Hospital (MGH) using the ECAT EXACT HR+ scanner (Siemens). 18F-flortaucipir (FTP) PET was introduced into HABS mid-study to measure tau burden, with participants undergoing their first FTP PET at 2.1 ± 1.7 years after the baseline visit. Longitudinal tau PET measurements were obtained at Year 1 (for those enrolled after FTP PET was introduced), 4, 6 and 9 visits. Detailed Aβ and tau PET protocols have been previously described.55 Aβ PET measurements were represented as a distribution volume ratio (DVR) across a composite of frontal, lateral temporal and parietal and retrosplenial regions (FLR), defined using FreeSurfer (version 6.0). Tau PET measurements were computed as standardized uptake value ratios (SUVR) within the inferior temporal cortex (ITC), a prominent site for early neocortical tau accumulation in preclinical AD56 which has been associated with emerging cognitive impairment.55 Both Aβ and tau PET used cerebellar grey matter as the reference region and were corrected for partial volume effects (PVC) using the geometric transfer matrix method.57
MRI
Brain MRI was obtained on Siemens Tim Trio 3-T scanner with a 12-channel phased-array head coil at MGH (Athinoula A. Martinos Center for Biomedical Imaging),45 at Years 1, 4, 6 and 9 visits. High-resolution 3D T1-weighted multi-echo magnetization-prepared, rapid acquisition gradient echo (MPRAGE) anatomical images were collected with the following parameters: repetition time (TR) = 2200 ms; multi-echo times (TE) = 1.54 ms, 3.36 ms, 5.18 and 7 ms; dimensions = 1.2 × 1.2 × 1.2 mm. MPRAGE images were processed with FreeSurfer longitudinal processing stream.58 Bilateral hippocampal volume was used as the primary neuroimaging marker of longitudinal neurodegeneration, with adjustment for total intracranial volume.59 For baseline white matter hyperintensity (WMH), 3D fluid-attenuated inversion recovery (FLAIR) images were collected with the following parameters: TR/TE/inversion time (TI) = 6000/454/2100 ms; dimensions = 1 × 1 × 1.5 mm. We segmented WMH on FLAIR images using the HyperMapp3r algorithm.60 HyperMapp3r is a convolutional neural network-based segmentation algorithm that uses T1-weighted, FLAIR and brain mask images to generate WMH predictions in the subject space. WMH volumes were log10-transformed prior to analysis.
Cognitive assessment
Cognition was assessed annually using the Preclinical Alzheimer Cognitive Composite-5 (PACC5).61 The PACC5 is a composite that includes the MMSE,50 Wechsler Adult Intelligence Scale–Revised Digit Symbol Coding,62 Wechsler Memory Scale–Revised Logical Memory delayed recall,51 Free and Cued Selective Reminding Test (free recall plus total recall)63 and Category Fluency Test.64
Cardiovascular risk
We calculated the office-based Framingham Heart Study Cardiovascular Disease risk score (FHS-CVD),65 which represents a sex-specific weighted sum of age, antihypertensive treatment (dichotomous), systolic blood pressure (SBP, mmHg), body mass index (BMI), diabetes status (dichotomous) and current cigarette smoking status (dichotomous). The FHS-CVD score provides a 10-year probability of future cardiovascular events, including coronary death, myocardial infarction, coronary insufficiency, angina, ischaemic stroke, haemorrhagic stroke, transient ischaemic attack, peripheral artery disease and heart failure.
Statistical analyses
We used R version 4.2.2 for statistical analyses. All continuous variables were z-transformed prior to analysis. We first used linear mixed effects model (‘nlme’ package) to examine the interactive effects of baseline plasma VEGF markers and Aβ burden (modelled as continuous variables) on PACC5 performance over time. Time was operationalized as years from baseline assessment. We adjusted for age, sex, years of education, their interactions with time, and included random intercepts and slopes. There were no significant effects of APOE ε4 carrier status or its interaction with time, and, accordingly, APOE ε4 was not included the final models. For secondary analysis, we examined the effects on longitudinal CDR Sum of Boxes (CDR-SOB) score as a measure of clinical progression and functional decline. For VEGF markers that showed no significant interaction with Aβ on longitudinal PACC5, we ran separate models without the interaction term to examine independent effects of VEGF markers and Aβ on PACC5 decline, adjusting for the same covariates as above. We applied Bonferroni correction for multiple comparisons (five VEGF measures), with P < 0.01 for significance.
We further examined whether any observed VEGF by Aβ interaction on longitudinal cognitive decline were mediated by other markers of AD pathology. We employed linear mixed effects models to examine the interactive effects of baseline plasma VEGF markers and Aβ burden (modelled as continuous variables) on (i) ITC tau PET burden; and (ii) hippocampal volume over time. Models were adjusted for age, sex, their interactions with time and included random intercepts and slopes. We applied Bonferroni correction for multiple comparisons (two VEGF measures), with P < 0.025 for significance.
We further used moderated mediation analysis (‘mediation’ package66) to examine whether tau accumulation mediated the interactive effects of VEGF markers and Aβ on PACC5 decline. Individual slopes of ITC tau changes and PACC5 changes were extracted from unadjusted linear mixed models with time as the only predictor, including both random slopes and intercepts. We conducted moderated mediation analysis with PACC5 slope as outcome, ITC tau slope as mediator, baseline Aβ burden (continuous) as moderator and adjusted for age, sex, years of education and time interval between baseline Aβ and first tau scan. To examine for moderation effects by baseline Aβ burden, mediation models were run at both low and high levels of baseline Aβ burden, defined respectively by the mean PiB burden of Aβ-negative (PiB PVC DVR = 1.17) and Aβ-positive (PiB PVC DVR = 1.84) participants, using the conventional Aβ-positivity threshold (PiB PVC DVR of 1.32 in HABS) for dichotomization.
Lastly, we examined whether any observed VEGF effects on longitudinal imaging measures or cognition may be related to FHS-CVD score or WMH. We examined the associations between VEGF measures and baseline FHS-CVD score or WMH, adjusting for age, sex and intracranial volume (for WMH only). We also repeated mixed effects models (ITC tau ∼ Plasma marker × Aβ × Time + covariates; and PACC5 ∼ Plasma marker × Aβ × Time + covariates) and additionally adjusted for FHS-CVD score or WMH and their interactions with time.
Results
Baseline characteristics of the study participants, including mean and range of follow-up duration, are summarized in Table 1.
Participant characteristics
| Characteristic, n = 317 except when specified | |
|---|---|
| Age at baseline, years, mean (SD) | 71.7 (7.9) |
| Females, n (%) | 194 (61) |
| Education, years, mean (SD) | 15.9 (2.9) |
| APOE ε4 carriers, n (%) | 89 (28) |
| Baseline PiB PET FLR DVR, PVC, mean (SD) | 1.37 (0.4) |
| Amyloid-β-positive, n (%) | 90 (28) |
| Baseline ITC tau PET SUVR, PVC, mean (SD), n = 187 | 1.44 (0.2) |
| No. of longitudinal tau PET scans, mean (SD; range), n = 187 | 2.6 (0.6; 2 to 4) |
| Duration of tau PET follow-up, years, mean (SD; range), n = 187 | 4.3 (1.6; 1.3 to 9) |
| No. of longitudinal PACC5 assessments, mean (SD; range) | 7.4 (2.7; 2 to 12) |
| Duration of cognitive follow-up, years, mean (SD; range) | 7.4 (3.2; 0.8 to 12.3) |
| Baseline hippocampal volume, mm3, mean (SD), n = 207 | 7228 (763) |
| No. of longitudinal MRIs, mean (SD; range), n = 207 | 3.3 (1.0; 2 to 6) |
| Duration of MRI follow-up, yr, mean (SD; range) n = 207 | 4.8 (1.8; 1.3 to 10.3) |
| Baseline FHS-CVD risk score, mean (SD) | 28.9 (18.2) |
| Baseline white matter hyperintensity, mm3, mean (SD) | 6044 (9534) |
| Characteristic, n = 317 except when specified | |
|---|---|
| Age at baseline, years, mean (SD) | 71.7 (7.9) |
| Females, n (%) | 194 (61) |
| Education, years, mean (SD) | 15.9 (2.9) |
| APOE ε4 carriers, n (%) | 89 (28) |
| Baseline PiB PET FLR DVR, PVC, mean (SD) | 1.37 (0.4) |
| Amyloid-β-positive, n (%) | 90 (28) |
| Baseline ITC tau PET SUVR, PVC, mean (SD), n = 187 | 1.44 (0.2) |
| No. of longitudinal tau PET scans, mean (SD; range), n = 187 | 2.6 (0.6; 2 to 4) |
| Duration of tau PET follow-up, years, mean (SD; range), n = 187 | 4.3 (1.6; 1.3 to 9) |
| No. of longitudinal PACC5 assessments, mean (SD; range) | 7.4 (2.7; 2 to 12) |
| Duration of cognitive follow-up, years, mean (SD; range) | 7.4 (3.2; 0.8 to 12.3) |
| Baseline hippocampal volume, mm3, mean (SD), n = 207 | 7228 (763) |
| No. of longitudinal MRIs, mean (SD; range), n = 207 | 3.3 (1.0; 2 to 6) |
| Duration of MRI follow-up, yr, mean (SD; range) n = 207 | 4.8 (1.8; 1.3 to 10.3) |
| Baseline FHS-CVD risk score, mean (SD) | 28.9 (18.2) |
| Baseline white matter hyperintensity, mm3, mean (SD) | 6044 (9534) |
APOE ε4 = apolipoprotein E ε4 allele; DVR = distribution volume ratio; FHS-CVD = Framingham Heart Study Cardiovascular Disease risk score; FLR = frontal, lateral temporal and parietal and retrosplenial regional uptake; ITC = inferior temporal cortex; PACC5 = Preclinical Alzheimer’s Cognitive Composite-5; PiB = Pittsburgh compound B; PVC = partial volume corrected; SD = standard deviation; SUVR = standardized uptake value ratio.
Participant characteristics
| Characteristic, n = 317 except when specified | |
|---|---|
| Age at baseline, years, mean (SD) | 71.7 (7.9) |
| Females, n (%) | 194 (61) |
| Education, years, mean (SD) | 15.9 (2.9) |
| APOE ε4 carriers, n (%) | 89 (28) |
| Baseline PiB PET FLR DVR, PVC, mean (SD) | 1.37 (0.4) |
| Amyloid-β-positive, n (%) | 90 (28) |
| Baseline ITC tau PET SUVR, PVC, mean (SD), n = 187 | 1.44 (0.2) |
| No. of longitudinal tau PET scans, mean (SD; range), n = 187 | 2.6 (0.6; 2 to 4) |
| Duration of tau PET follow-up, years, mean (SD; range), n = 187 | 4.3 (1.6; 1.3 to 9) |
| No. of longitudinal PACC5 assessments, mean (SD; range) | 7.4 (2.7; 2 to 12) |
| Duration of cognitive follow-up, years, mean (SD; range) | 7.4 (3.2; 0.8 to 12.3) |
| Baseline hippocampal volume, mm3, mean (SD), n = 207 | 7228 (763) |
| No. of longitudinal MRIs, mean (SD; range), n = 207 | 3.3 (1.0; 2 to 6) |
| Duration of MRI follow-up, yr, mean (SD; range) n = 207 | 4.8 (1.8; 1.3 to 10.3) |
| Baseline FHS-CVD risk score, mean (SD) | 28.9 (18.2) |
| Baseline white matter hyperintensity, mm3, mean (SD) | 6044 (9534) |
| Characteristic, n = 317 except when specified | |
|---|---|
| Age at baseline, years, mean (SD) | 71.7 (7.9) |
| Females, n (%) | 194 (61) |
| Education, years, mean (SD) | 15.9 (2.9) |
| APOE ε4 carriers, n (%) | 89 (28) |
| Baseline PiB PET FLR DVR, PVC, mean (SD) | 1.37 (0.4) |
| Amyloid-β-positive, n (%) | 90 (28) |
| Baseline ITC tau PET SUVR, PVC, mean (SD), n = 187 | 1.44 (0.2) |
| No. of longitudinal tau PET scans, mean (SD; range), n = 187 | 2.6 (0.6; 2 to 4) |
| Duration of tau PET follow-up, years, mean (SD; range), n = 187 | 4.3 (1.6; 1.3 to 9) |
| No. of longitudinal PACC5 assessments, mean (SD; range) | 7.4 (2.7; 2 to 12) |
| Duration of cognitive follow-up, years, mean (SD; range) | 7.4 (3.2; 0.8 to 12.3) |
| Baseline hippocampal volume, mm3, mean (SD), n = 207 | 7228 (763) |
| No. of longitudinal MRIs, mean (SD; range), n = 207 | 3.3 (1.0; 2 to 6) |
| Duration of MRI follow-up, yr, mean (SD; range) n = 207 | 4.8 (1.8; 1.3 to 10.3) |
| Baseline FHS-CVD risk score, mean (SD) | 28.9 (18.2) |
| Baseline white matter hyperintensity, mm3, mean (SD) | 6044 (9534) |
APOE ε4 = apolipoprotein E ε4 allele; DVR = distribution volume ratio; FHS-CVD = Framingham Heart Study Cardiovascular Disease risk score; FLR = frontal, lateral temporal and parietal and retrosplenial regional uptake; ITC = inferior temporal cortex; PACC5 = Preclinical Alzheimer’s Cognitive Composite-5; PiB = Pittsburgh compound B; PVC = partial volume corrected; SD = standard deviation; SUVR = standardized uptake value ratio.
We first investigated whether baseline plasma VEGF proteins have interactive effects with baseline Aβ PET burden on prospective cognitive decline in CU older adults. Of the five VEGF proteins, VEGFA and PGF showed significant but opposing interactions with Aβ on longitudinal PACC5 decline. Specifically, low VEGFA and high PGF were separately associated with accelerated cognitive decline in individuals with elevated baseline Aβ [VEGFA × Aβ × Time: β = 0.06 (0.03 to 0.10), t = 3.25, P = 0.001; PGF × Aβ × Time: β = −0.08 (−0.12 to −0.04), t = −3.91, P < 0.001]. Figure 1 visualizes the model results (where Aβ and VEGF levels were modelled continuously) by presenting estimated PACC5 trajectories based on different levels of baseline Aβ and VEGFA/PGF. The individual (unadjusted) PACC5 trajectories are shown in Supplementary Fig. 1. There were no main effects of VEGFA or PGF on longitudinal cognition beyond their interactions with Aβ [VEGFA × Time: β = 0.01 (−0.04 to 0.06), t = 0.35, P = 0.73; PGF × Time: β = −0.02 (−0.07 to 0.03), t = −0.71 P = 0.48]. Including both VEGFA and PGF in the same regression model revealed essentially unchanged effect size of their interactions with Aβ on longitudinal cognition [VEGFA × Aβ × Time: β = 0.06 (0.02 to 0.10), t = 3.23, P = 0.001; PGF × Aβ × Time: β = −0.08 (−0.12 to −0.04), t = −3.88, P < 0.001], suggesting they have independent effects on Aβ-related cognitive decline. Secondary analyses examining longitudinal CDR-SOB score as a measure of clinical/functional progression revealed significant VEGFA–Aβ and PGF–Aβ interactions in directions concordant with their effects on PACC5. Specifically, low VEGFA and high PGF in baseline were associated with greater longitudinal progression on CDR-SOB in individuals with elevated baseline Aβ [VEGFA × Aβ × Time: β = −0.07 (−0.02 to −0.13), t = −2.59, P = 0.01; PGF × Aβ × Time: β = 0.10 (0.04 to 0.16), t = 3.21, P = 0.001; Fig. 2]. The remaining plasma VEGF markers (soluble FLT1, VEGFC and VEGFD) showed no significant effects on longitudinal cognitive decline, alone or interactively with Aβ (Supplementary Table 1).
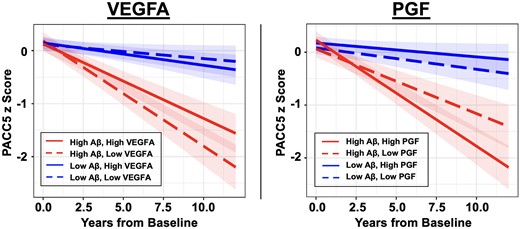
Effects of plasma VEGFA and PGF on cognitive decline. Linear mixed effects models revealed significant interactions between baseline amyloid-β (Aβ) burden and plasma VEGFA/PGF on longitudinal cognitive decline measured with the Preclinical Alzheimer’s Cognitive Composite-5 (PACC5). Individuals with elevated baseline Aβ and low VEGFA (left, dashed red line) or high PGF (right, solid red line) showed faster PACC5 decline. Baseline Aβ burden and VEGFA/PGF levels were modelled as continuous variables. To visualize the model results, estimated PACC5 trajectories based on different levels of baseline Aβ burden and VEGFA/PGF are presented. Low and high plasma marker levels are represented by −1 and +1 standard deviations relative to the mean. Low and high Aβ are represented by the mean Pittsburgh compound-B distribution volume ratio (PiB DVR) of amyloid-negative (1.17) and amyloid-positive (1.84) participants, respectively, dichotomized using the conventional amyloid-positivity threshold (PiB PVC-DVR of 1.32) in the Harvard Aging Brain Study (HABS) cohort for illustration purpose.
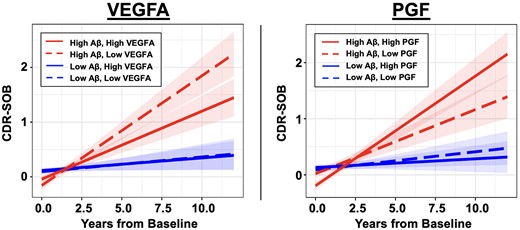
Effects of plasma VEGFA and PGF on progression of Clinical Dementia Rating Sum of Boxes score. Linear mixed effects models revealed significant interactions between baseline amyloid-β (Aβ) burden and plasma VEGFA/PGF on clinical progression measured with longitudinal Clinical Dementia Rating Sum of Boxes (CDR-SOB) score. Individuals with elevated baseline Aβ and low VEGFA (left, dashed red line) or high PGF (right, solid red line) showed faster progression in CDR-SOB. Baseline Aβ burden and VEGFA/PGF levels were modelled as continuous variables. To visualize the model results, estimated CDR-SOB trajectories based on different levels of baseline Aβ burden and VEGFA/PGF are presented. Low and high plasma markers are represented by −1 and +1 standard deviations relative to the mean. Low and high Aβ are represented by the mean Pittsburgh compound-B distribution volume ratio (PiB DVR) of amyloid-negative (1.17) and amyloid-positive (1.84) participants respectively, dichotomized using the conventional amyloid-positivity threshold (PiB PVC-DVR of 1.32) in the Harvard Aging Brain Study (HABS) cohort for illustration purpose.
We next examined whether the effects of VEGFA and PGF on cognitive decline may be mediated by other pathologies in the AD cascade, specifically tau accumulation and hippocampal atrophy. Both VEGFA and PGF demonstrated significant interactive effects with baseline Aβ burden on longitudinal ITC tau accumulation, in a direction concordant with their effects on cognitive decline. Low VEGFA and high PGF were associated with accelerated ITC tau accumulation when baseline Aβ was elevated [VEGFA × Aβ × Time: β = −0.12 (−0.17 to −0.07), t = −4.62, P < 0.001; PGF × Aβ × Time: β = 0.09 (0.04 to 0.14), t = 3.64, P < 0.001]. Additionally, there was a main effect of low VEGFA on greater ITC tau accumulation [VEGFA × Time: β = −0.08 (−0.13 to −0.03), t = −4.52, P < 0.001]. In contrast, there was no main effect of PGF on longitudinal ITC tau beyond its interaction with Aβ [PGF × Time: β = −0.02 (−0.03 to 0.08), t = 0.75, P = 0.45]. Figure 3 visualizes the model results (where Aβ and VEGF levels were modelled continuously) by presenting estimated ITC tau trajectories based on different levels of baseline Aβ and VEGFA/PGF. The individual (unadjusted) ITC tau trajectories are shown in Supplementary Fig. 2. We then ran separate moderated mediation models to formally examine whether ITC tau change significantly mediated the interaction between VEGFA or PGF with Aβ on cognitive decline. Results demonstrated that in individuals with elevated baseline Aβ burden, faster ITC tau accumulation fully mediated the detrimental effects of low VEGFA [β = 0.61 (0.34 to 0.90), P < 0.001] and partially mediated the detrimental effect of high PGF [β = −0.34 (−0.58 to −0.10), P = 0.003, proportion mediated = 31%] on accelerating cognitive decline (Fig. 4). In individuals with low baseline Aβ burden, there was no significant total or mediated effects of VEGFA or PGF on cognitive decline.
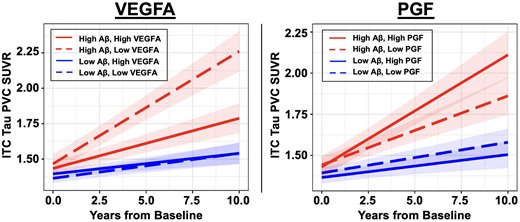
Effects of plasma VEGFA and PGF on longitudinal tau PET burden. Linear mixed effects models revealed significant interactions between baseline amyloid-β (Aβ) burden and plasma VEGFA/PGF on longitudinal tau pathology in inferior temporal cortex (ITC). Individuals with elevated baseline Aβ burden and low VEGFA (left, dashed red line) or high PGF (right, solid red line) showed accelerated ITC tau accumulation. Baseline Aβ burden and VEGFA/PGF levels were modelled as continuous variables. To visualize the model results, estimated ITC tau trajectories based on different levels of baseline Aβ burden and VEGFA/PGF are presented. Low and high plasma markers are represented by −1 and +1 standard deviations relative to the mean. Low and high Aβ is represented by the mean Pittsburgh compound-B distribution volume ratio (PiB DVR) of amyloid-negative (1.17) and amyloid-positive (1.84) participants respectively, dichotomized using the conventional amyloid-positivity threshold (PiB PVC-DVR of 1.32) in the Harvard Aging Brain Study (HABS) cohort for illustration purpose. SUVR = standardized uptake value ratio.
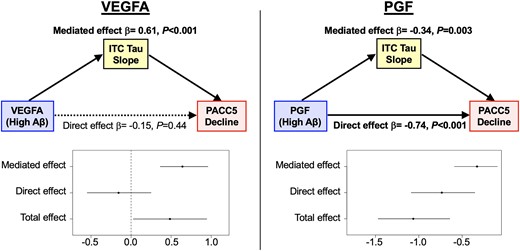
Tau mediates the effects of low VEGFA and high PGF on faster cognitive decline in preclinical Alzheimer’s disease. Results from moderated mediation analyses demonstrated that at an elevated level of baseline amyloid-β (Aβ) burden, accelerated accumulation of inferior temporal cortex (ITC) tau pathology fully mediated the effects of low VEGFA and partially mediated (31%) the effects of high PGF on faster Preclinical Alzheimer’s Cognitive Composite-5 (PACC5) decline. Elevated Aβ burden was defined using the mean Pittsburgh compound-B partial volume corrected distribution volume ratio (PiB PVC-DVR) burden of Aβ-positive participants in the Harvard Aging Brain Study (HABS), which was 1.84.
In contrast to the significant effects on longitudinal tau, low VEGFA only showed nominally significant interactive effects with baseline Aβ in association with faster longitudinal hippocampal atrophy [β = 0.02 (0.00 to 0.03), t = 1.97, P = 0.05] but did not survive correction for multiple comparisons. Similarly, high PGF showed non-significant but concordant interactive effects with baseline Aβ on greater hippocampal atrophy [β = −0.01 (−0.03 to 0.01), t = −1.08, P = 0.28]. In addition, high PGF was independently associated with smaller baseline hippocampal volume [β = −0.17 (−0.31 to −0.04), t = −2.56, P = 0.01].
Lastly, we examined whether the effects of VEGFA and PGF on tau and cognitive decline were distinct from other common measures of vascular risk or injury, specifically FHS-CVD score and WMH. There were no significant associations between baseline VEGFA or PGF with FHS-CVD score (P > 0.22) or WMH (P > 0.08), adjusting for age, sex and (for WMH only) intracranial volume. In addition, the interactive effects of VEGFA and PGF with Aβ burden on longitudinal ITC tau accumulation and PACC5 decline remain significant after adjusting for baseline FHS-CVD score or WMH (Table 2).
Summary of linear mixed effects models adjusting for markers of systemic vascular risk and cerebrovascular injury
| Adjusting for FHS-CVD × Time | Adjusting for WMH × Time | |||||
|---|---|---|---|---|---|---|
| β Estimate (95% CI) | t-value | P-value | β Estimate (95% CI) | t-value | P-value | |
| ITC tau | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | −0.10 (−0.15 to −0.04) | −3.72 | <0.001 | −0.15 (−0.21 to −0.10) | −5.36 | <0.001 |
| VEGFA × Time | −0.08 (−0.13 to −0.03) | −3.16 | 0.002 | −0.09 (−0.14 to −0.04) | −3.32 | 0.001 |
| PGF model | ||||||
| PGF × Aβ × Time | 0.09 (0.04 to 0.13) | 3.79 | <0.001 | 0.08 (0.03 to 0.14) | 3.33 | 0.001 |
| PGF × Time | 0.03 (−0.02 to 0.09) | 1.18 | 0.24 | 0.02 (−0.04 to 0.09) | 0.77 | 0.44 |
| PACC5 | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | 0.05 (0.02 to 0.09) | 2.83 | 0.005 | 0.07 (0.03 to 0.11) | 3.57 | <0.001 |
| VEGFA × Time | 0.01 (−0.04 to 0.05) | 0.29 | 0.77 | 0.01 (−0.04 to 0.06) | 0.24 | 0.81 |
| PGF model | ||||||
| PGF × Aβ × Time | −0.08 (−0.12 to −0.04) | −4.06 | <0.001 | −0.07 (−0.12 to −0.03) | −3.35 | <0.001 |
| PGF × Time | −0.04 (−0.09 to 0.01) | −1.40 | 0.16 | −0.07 (−0.01 to 0.05) | −0.36 | 0.72 |
| Adjusting for FHS-CVD × Time | Adjusting for WMH × Time | |||||
|---|---|---|---|---|---|---|
| β Estimate (95% CI) | t-value | P-value | β Estimate (95% CI) | t-value | P-value | |
| ITC tau | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | −0.10 (−0.15 to −0.04) | −3.72 | <0.001 | −0.15 (−0.21 to −0.10) | −5.36 | <0.001 |
| VEGFA × Time | −0.08 (−0.13 to −0.03) | −3.16 | 0.002 | −0.09 (−0.14 to −0.04) | −3.32 | 0.001 |
| PGF model | ||||||
| PGF × Aβ × Time | 0.09 (0.04 to 0.13) | 3.79 | <0.001 | 0.08 (0.03 to 0.14) | 3.33 | 0.001 |
| PGF × Time | 0.03 (−0.02 to 0.09) | 1.18 | 0.24 | 0.02 (−0.04 to 0.09) | 0.77 | 0.44 |
| PACC5 | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | 0.05 (0.02 to 0.09) | 2.83 | 0.005 | 0.07 (0.03 to 0.11) | 3.57 | <0.001 |
| VEGFA × Time | 0.01 (−0.04 to 0.05) | 0.29 | 0.77 | 0.01 (−0.04 to 0.06) | 0.24 | 0.81 |
| PGF model | ||||||
| PGF × Aβ × Time | −0.08 (−0.12 to −0.04) | −4.06 | <0.001 | −0.07 (−0.12 to −0.03) | −3.35 | <0.001 |
| PGF × Time | −0.04 (−0.09 to 0.01) | −1.40 | 0.16 | −0.07 (−0.01 to 0.05) | −0.36 | 0.72 |
The effects of low VEGFA and high PGF on accelerating amyloid-related inferior temporal cortex (ITC) tau accumulation and Preclinical Alzheimer’s Cognitive Composite-5 (PACC5) decline remained significant after adjusting for baseline Framingham Heart Study Cardiovascular Disease risk (FHS-CVD) score and white matter hyperintensity (WMH) volume. Aβ = β-amyloid; CI = confidence interval.
Summary of linear mixed effects models adjusting for markers of systemic vascular risk and cerebrovascular injury
| Adjusting for FHS-CVD × Time | Adjusting for WMH × Time | |||||
|---|---|---|---|---|---|---|
| β Estimate (95% CI) | t-value | P-value | β Estimate (95% CI) | t-value | P-value | |
| ITC tau | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | −0.10 (−0.15 to −0.04) | −3.72 | <0.001 | −0.15 (−0.21 to −0.10) | −5.36 | <0.001 |
| VEGFA × Time | −0.08 (−0.13 to −0.03) | −3.16 | 0.002 | −0.09 (−0.14 to −0.04) | −3.32 | 0.001 |
| PGF model | ||||||
| PGF × Aβ × Time | 0.09 (0.04 to 0.13) | 3.79 | <0.001 | 0.08 (0.03 to 0.14) | 3.33 | 0.001 |
| PGF × Time | 0.03 (−0.02 to 0.09) | 1.18 | 0.24 | 0.02 (−0.04 to 0.09) | 0.77 | 0.44 |
| PACC5 | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | 0.05 (0.02 to 0.09) | 2.83 | 0.005 | 0.07 (0.03 to 0.11) | 3.57 | <0.001 |
| VEGFA × Time | 0.01 (−0.04 to 0.05) | 0.29 | 0.77 | 0.01 (−0.04 to 0.06) | 0.24 | 0.81 |
| PGF model | ||||||
| PGF × Aβ × Time | −0.08 (−0.12 to −0.04) | −4.06 | <0.001 | −0.07 (−0.12 to −0.03) | −3.35 | <0.001 |
| PGF × Time | −0.04 (−0.09 to 0.01) | −1.40 | 0.16 | −0.07 (−0.01 to 0.05) | −0.36 | 0.72 |
| Adjusting for FHS-CVD × Time | Adjusting for WMH × Time | |||||
|---|---|---|---|---|---|---|
| β Estimate (95% CI) | t-value | P-value | β Estimate (95% CI) | t-value | P-value | |
| ITC tau | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | −0.10 (−0.15 to −0.04) | −3.72 | <0.001 | −0.15 (−0.21 to −0.10) | −5.36 | <0.001 |
| VEGFA × Time | −0.08 (−0.13 to −0.03) | −3.16 | 0.002 | −0.09 (−0.14 to −0.04) | −3.32 | 0.001 |
| PGF model | ||||||
| PGF × Aβ × Time | 0.09 (0.04 to 0.13) | 3.79 | <0.001 | 0.08 (0.03 to 0.14) | 3.33 | 0.001 |
| PGF × Time | 0.03 (−0.02 to 0.09) | 1.18 | 0.24 | 0.02 (−0.04 to 0.09) | 0.77 | 0.44 |
| PACC5 | ||||||
| VEGFA model | ||||||
| VEGFA × Aβ × Time | 0.05 (0.02 to 0.09) | 2.83 | 0.005 | 0.07 (0.03 to 0.11) | 3.57 | <0.001 |
| VEGFA × Time | 0.01 (−0.04 to 0.05) | 0.29 | 0.77 | 0.01 (−0.04 to 0.06) | 0.24 | 0.81 |
| PGF model | ||||||
| PGF × Aβ × Time | −0.08 (−0.12 to −0.04) | −4.06 | <0.001 | −0.07 (−0.12 to −0.03) | −3.35 | <0.001 |
| PGF × Time | −0.04 (−0.09 to 0.01) | −1.40 | 0.16 | −0.07 (−0.01 to 0.05) | −0.36 | 0.72 |
The effects of low VEGFA and high PGF on accelerating amyloid-related inferior temporal cortex (ITC) tau accumulation and Preclinical Alzheimer’s Cognitive Composite-5 (PACC5) decline remained significant after adjusting for baseline Framingham Heart Study Cardiovascular Disease risk (FHS-CVD) score and white matter hyperintensity (WMH) volume. Aβ = β-amyloid; CI = confidence interval.
Discussion
In a well-characterized cohort of 317 CU older adults followed with longitudinal multimodal neuroimaging and up to 12 years of annual cognitive follow-up, we examined the interactive and independent effects of plasma VEGF proteins and Aβ PET burden on prospective cognitive decline and further determined the association with other markers of AD and vascular pathologies. We demonstrated that low VEGFA and high PGF in baseline plasma were synergistic with elevated Aβ burden in driving faster cognitive decline, which was corroborated by a consistent effect on faster CDR-SOB progression. Concordantly, low VEGFA and high PGF were associated with accelerated longitudinal ITC tau accumulation in individuals with elevated Aβ. Moderated mediation analyses confirmed that accelerated tau accumulation fully mediated the effects of low VEGFA and partially mediated the effects of high PGF (31%) on faster cognitive decline. Importantly, the effects of VEGFA and PGF on tau and cognition were distinct from conventional vascular risk and injury measures. To our knowledge, this is the first study to examine the effects of plasma VEGF proteins on longitudinal tau pathology and cognitive decline in preclinical AD. Our results highlight VEGFA and PGF as potential molecular targets, in combination with anti-Aβ and traditional vascular risk reduction therapies, to modify the early vascular-AD synergy and delay or ideally prevent the onset of cognitive decline.
VEGFA was the first-identified VEGF ligand and remains the most-studied member of the VEGF protein family. It is traditionally viewed as a canonical pro-angiogenesis factor, though newer research has revealed multifaceted functions in vascular tone, neuroprotection and blood–brain barrier permeability.17-20 Our results support a protective role of VEGFA in slowing cognitive decline in preclinical AD. This is consistent with prior studies demonstrating associations between higher CSF VEGFA and better cognitive function in AD.24,26,31,35 In particular, one study demonstrated a concordant VEGFA by Aβ interaction where higher CSF VEGFA was protective against longitudinal Aβ-related cognitive decline in a cohort of CU, MCI and AD participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI).35 Our current study expands upon these findings and supports high plasma VEGFA, a notably less invasive and more broadly applicable biomarker, is similarly associated with slower Aβ-related cognitive decline in a CU-only cohort. This highlights the utility of plasma VEGFA to inform the risk of progression in preclinical AD. Importantly, we found that the protective effects of VEGFA on Aβ-related cognitive decline were fully explained by a slower rate of tau accumulation. This novel finding provides strong support for a mechanistic role of VEGFA in the early pathogenesis of AD.
The mechanisms through which VEGFA protects against Aβ-associated tau accumulation and cognitive decline require further investigation. Cerebral hypoperfusion is a well-recognized feature in AD and has been associated with increased tau pathology in animal67,68 and human69,70 studies. Endothelin, the major vasoconstrictor in cerebral vasculature, has been shown to be upregulated by Aβ in neuronal and endothelial cells and is elevated in AD brain issue and leptomeningeal blood vessels.71,72 In this context, VEGFA stimulation of nitric oxide release, endothelial relaxation and vasodilation73 may offset the vasoconstriction and hypoperfusion induced by Aβ. Indeed, increased levels of VEGFA and VEGF receptor 2 (its major signalling partner) were associated with restoration of normoperfusion in an AD mouse model without evidence of increased angiogenesis.74 Alternatively, VEGFA has demonstrated neuroprotective effects in face of various stressors18 and has been shown to rescue synaptic dysfunction associated with oligomeric Aβ in an in vitro model.75 Lastly, as VEGFA binds tightly to Aβ in vitro and colocalizes with neuritic plaques in post-mortem brain tissue of patients with AD,76 higher levels of VEGFA may help overcome this sequestration effect in the presence of Aβ.35
In contrast to VEGFA, we found that higher plasma PGF was independently associated with accelerated cognitive decline in those with preclinical AD. Our findings are consistent with prior studies in mixed clinical cohorts demonstrating higher plasma PGF was associated with increased risk of functional cognitive impairment48 and that higher PGF expression in prefrontal cortex and whole blood was associated with faster cognitive decline.30,47 Interestingly, our results indicated that roughly only one-third of the effects of PGF on Aβ-related cognitive decline were mediated by accelerated early neocortical tau pathology, suggesting a partially distinct mechanism from VEGFA, which was fully mediated by tau. PGF signals via membrane-bound VEGF receptor 1 (or FLT1) and is implicated in pathological angiogenesis and inflammation.20 PGF and FLT1 are involved in monocyte and macrophage migration to the site of injury and increased inflammatory cytokine production.77,78 Another study further suggests that FLT1 signalling is required for microglial chemotaxis in response to Aβ.79 Therefore, PGF may exacerbate Aβ-related cognitive decline by promoting neuroinflammation through recruiting microglia and peripherally-derived macrophages to Aβ plaques, which has been shown to promote tau phosphorylation and accumulation.80,81
We recently identified synergistic interactions between high systemic cardiovascular risk and Aβ burden on accelerating tau accumulation and cognitive decline in preclinical AD.15 In this context, we examined whether VEGF markers have independent effects from conventional measures of cardiovascular risk (FHS-CVD score) or cerebrovascular injury (WMH). We found no significant associations between plasma VEGF measures with FHS-CVD or WMH. Additionally, the effects of VEGFA and PGF remained significant after adjusting for FHS-CVD score or WMH. Our findings are discordant from a recent study demonstrating higher plasma PGF was associated with greater cross-sectional WMH in a cohort of CU and impaired individuals with varying levels of vascular risk.48 In addition, Gertje et al.82 found greater CSF PGF was associated with higher WMH in a CU-only cohort, with or without adjusting for Aβ-pathology. Our divergent finding may be related to measurement of peripheral PGF (in contrast to CSF PGF in Gertje et al.82) and cohort differences—we included only CU older adults from the community, while Gertje et al.82 also included memory clinic patients with subjective cognitive decline but normal cognitive testing who may have slightly more advanced pathology. Taken together, our findings supports that in community dwelling CU adults with an overall average level of cardiovascular risk, VEGFA and PGF appear to capture distinct underlying processes from systemic vascular risks and WM microvascular disease in driving Aβ-related tau pathology and cognitive decline, highlighting their potential as new combinational therapeutic targets.
In this context, anti-PGF therapy may be an attractive approach. The expression of PGF is typically low under physiological conditions but undergoes significant upregulation in pathological states.83 Importantly, in vitro and animal studies have shown that anti-PGF treatment reduced inflammatory infiltrates in atherosclerotic plaques and inhibited pathological angiogenesis without impacting healthy vessels,84,85 highlighting the feasibility of PGF as a novel therapeutic target. Direct modulation of VEGFA will require further investigation, given its widespread physiological functions and concern that dysregulated VEGFA signalling may contribute to increased capillary stalling and blood–brain barrier permeability in more advanced AD.41,42,86 However, there may be other strategies to safely augment VEGFA in preclinical AD. For example, exercise training has been shown to upregulate VEGFA, which rescued age-related hypoperfusion87 and Aβ-related spatial learning and memory deficits88 in animal models and improve peripheral vascular function in AD patients.89
We did not observe a significant interaction between VEGFA or PGF and Aβ on accelerated hippocampal atrophy, although the directions of association were aligned with the effects on tau and cognition. Our findings diverge from a prior study34 using the ADNI cohort (including CU, MCI and AD participants) which showed a protective effect of higher CSF VEGFA on reducing hippocampal and global atrophy, in individuals who were CSF Aβ-positive but not in those who were Aβ-negative. However, another study35 using data from ADNI did not observe a significant interaction between CSF VEGFA and Aβ (used as continuous measure as in the current study) on longitudinal hippocampal volume. It may be that the power to capture significantly divergent neurodegeneration, a later event in the AD cascade, is further diminished in our preclinical sample, in addition to using an interaction model.
The current study has several limitations. First, we did not have CSF measures of VEGF proteins to complement our peripheral measurements. Future studies are needed to confirm the effects of CSF VEGFA and PGF in preclinical AD. Similarly, as we measured circulating proteins, we were unable to examine membrane-bound FLT1 (the major signalling partner of PGF) which was shown in clinicopathological studies to be associated with faster cognitive decline and greater AD neuropathology.30,46 In addition, VEGFB, another high-affinity ligand for FTL1, was not available on the immunoassay kit used in our study and was therefore not evaluated. Given that brain VEGFB expression has been associated with greater AD neuropathology and cognitive decline30,46 the predictive utility of plasma VEGFB should be assessed in future studies. Lastly, the HABS cohort consists of highly educated, predominantly non-Hispanic white individuals and excluded those with symptomatic cerebrovascular disease and poorly controlled vascular risk factors; this may limit the generalizability of our findings to other populations with diverse racial background or very high levels of vascular risks.
In summary, our study leveraged a large, well-characterized prospective CU cohort with one of the longest neuroimaging and cognitive follow up to date to address the mechanisms by which early vascular dysfunction may promote cognitive decline in preclinical AD. We demonstrated that low VEGFA and high PGF in plasma have synergistic effects with elevated Aβ in driving faster cognitive decline, which was significantly mediated by accelerated early neocortical tau accumulation. The results here expand on previous CSF studies and support the potential utility of minimally-invasive plasma VEGFA and PGF measures in clinical and translational research studies of the earliest stages of AD. Importantly, VEGFA and PGF appear to capture biological mechanisms distinct from conventional vascular risk and cerebrovascular injury. Taken together, our study supports the potential of VEGFA and PGF as new therapeutic targets, in combination with anti-Aβ and vascular risk reduction strategies, to meaningfully modify the trajectory of preclinical AD.
Data availability
Data from the Harvard Aging Brain Study can be requested at https://habs.mgh.harvard.edu/researchers/request-data. Longitudinal data will be made available in periodic data releases at the same web address or via direct request to the corresponding author(s). Qualified investigators must abide by the Harvard Aging Brain Study online data use agreement, designed to protect the privacy of our participants.
Acknowledgements
We thank the study participants of the Harvard Aging Brain Study.
Funding
This work was supported by the National Institutes of Health (NIH) [grant numbers P01 AG036694 (K.A.J. and R.A.S.); K23 AG049087 and R01 AG062667 (J.P.C.); K23 AG062750 (H.-S.Y.); K24 AG035007 (R.A.S.); R01 AG061518 (T.J.H.); P30 AG062421 and R01 AG062306 (B.C.C. and S.E.A.)]; the American Academy of Neurology, American Brain Foundation and McKnight Brain Research Foundation [McKnight Clinical Translational Research Scholarship in Cognitive Aging and Age-Related Memory Loss] (W.-Y.W.Y.); and the Doris Duke Charitable Foundation Clinical Scientist Development Award (J.P.C.). This research was carried out in part at the Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies (CFNT), P41EB015896, a P41 Biotechnology Resource Grant supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program; specifically, grant numbers S10RR021110, S10RR023401 and S10RR023043. Funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Competing interests
The authors report no competing interests relevant to the current study. Potential competing interests outside of the submitted work are as follows. A.P.S. has served as a consultant for Janssen Pharmaceuticals and Biogen. D.M.R. has served as a consultant for Biogen Idec, Novartis and the Dana Foundation. J.P.C. has served as a consultant for Humana Healthcare, ExpertConnect and Leerink Partners. K.A.J. has served as a consultant for Merck and Novartis. R.A.S. has served as a consultant for AC Immune, Acumen, Genentech, Ionis, Janssen, Merck, Novartis, Oligomerix, Prothena, Renew, Shionogi and Vaxxinity. K.A.J. and R.A.S. are involved in public-private partnership clinical trials sponsored by the National Institutes of Health and Eli Lilly, which owns the distribution rights to flortaucipir, but they do not have any personal financial relationship with Eli Lilly. T.J.H. has served on the Scientific Advisory Board for Vivid Genomics.
Supplementary material
Supplementary material is available at Brain online.
References
Author notes
Hyun-Sik Yang and Wai-Ying Wendy Yau contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}