




This scientific commentary refers to ‘Neurophysiological markers of motor compensatory mechanisms in early Parkinson’s disease’ by Passaretti et al. (https://doi.org/10.1093/brain/awae210).
One of the most notable characteristics of Parkinson’s disease is the asymmetry of motor clinical symptoms that may be evident from disease onset in both sporadic as well as hereditary forms. Symptom-side predominance is likely a complex combination of the cascading effects of striatal degeneration, influenced by both genetically encoded mechanisms as well as acquired differences in the vulnerability of degenerating neurons and their projections.1 Several factors have been proposed to contribute to this asymmetrical development of motor symptoms, ranging from handedness and hemispheric dominance2 to the anatomical location of the initial α-synuclein pathology and its connectome.3 Yet, it remains unlikely that asymmetry of dopaminergic denervation and motor dysfunction can be fully accounted for by a single pathophysiological factor.4 This is illustrated by the possible decorrelation observed in patients with significant bilateral dopaminergic loss, but unilateral clinical symptoms. In this issue of Brain, Passaretti and colleagues5 focus on this group of patients to assess the potential role of motor compensation, i.e. the ability of the brain to compensate for the progressive loss of dopamine-producing neurons. This adaptation may be so effective that individuals may show no clinical signs of Parkinson’s disease despite substantial neurodegeneration (similar to an extended prodromal phase), or they may develop motor symptoms on only one side of the body even though the underlying dopaminergic deficit is bilateral. However, the mechanisms of motor compensation have proved difficult to identify and to disentangle from the increasing burden of neurodegeneration over the course of the disease.6
Using clinical evaluation and dopamine transporter imaging, Passaretti and colleagues5 screened 82 individuals with Parkinson’s disease and found that 16 had bilateral nigrostriatal degeneration but only unilateral motor symptoms. The authors then used transcranial magnetic stimulation to probe potential compensatory changes in response to the abnormal inputs that M1 receives from subcortical structures: increased corticospinal excitability, reduced intracortical inhibition, and increased M1 long-term potentiation. Finally, they developed a model based on clinical and imaging data to quantify motor compensation and its correlation with neurophysiological data (Fig. 1).
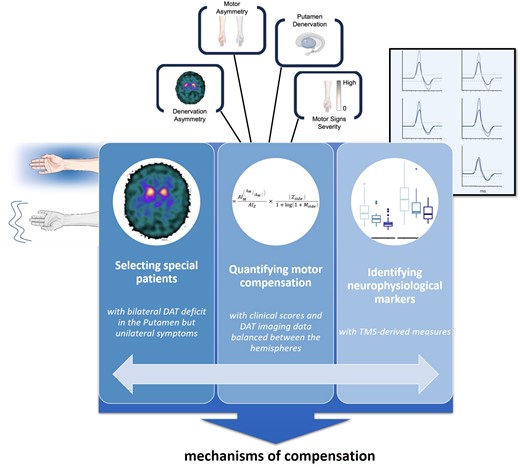
Passaretti and colleagues’ empirical approach to identifying neurophysiological markers of motor compensatory mechanisms in early Parkinson’s disease. Left: Patients with bilateral nigrostriatal degeneration but only unilateral motor symptoms—presumably as a result of motor compensation—were identified. Middle: A metric quantifying motor compensation was developed, based on the asymmetry of clinical and molecular imaging data. Right: Transcranial magnetic stimulation (TMS) was used to probe the neurophysiological underpinnings of adaptive changes in cortical excitability, plasticity and interhemispheric inhibition. This innovative approach pinpointed increased corticospinal excitability and plasticity as potential mechanisms of motor compensation, as well as reduced inhibition of the symptomatic hemisphere, related to lower putamen dopamine transporter (DAT) binding. Further studies in larger and more diverse cohorts are required to validate these findings, and to track the progression of compensatory mechanisms over time. Understanding these mechanisms could drive the development of targeted therapies aimed at enhancing the natural ability of the brain to cope with neurodegeneration. Illustrations modified from Figs 1–3 and 5 in Passaretti et al.5).
The model successfully discriminated the presymptomatic from the symptomatic hemisphere, and showed that motor compensation in the presymptomatic hemisphere is associated with increased plasticity. This phenomenon was found to depend on striatal dopamine levels, with greater degrees of denervation associated with increased plasticity in the presymptomatic hemisphere. Compensation for dopamine loss was thus interpreted by the authors as a ramping up of activity in certain areas to maintain correct motor function. This mechanism was observed in multiple brain regions including the sensorimotor cortex, putamen, caudate nuclei, and bilateral projections between hemispheres, and involved both activating and inhibitory outputs.
This work is particularly remarkable for the way in which it overcomes empirical obstacles to identifying compensatory mechanisms. First, it focuses on a very specific patient population that can shed light on compensatory mechanisms that would otherwise be overlooked. Second, it combines molecular imaging with various types of neurophysiological data, as well as an innovative new metric, to test the specific predictions of the compensatory hypothesis and align biological data with clinical observations. The authors interpret their data as suggesting that activity in brain areas involved in goal-directed motor tasks may compensate for the altered function of automatic motor networks. In simpler terms, brain areas responsible for complex motor tasks may draw upon their greater plasticity to perform activities that were previously automatic, adapting at the cost of increased resource consumption and more intense motor output. Although this study requires replication with patients at different stages of Parkinson’s disease, particularly in the prodromal phase, the implications of these original and ground-breaking data are substantial.
Understanding the dynamics of motor compensation provides clues to why significant dopaminergic denervation does not always result in the full spectrum of Parkinson’s disease symptoms. Passaretti and colleagues5 propose that affected brain regions operate in a finely tuned, albeit precariously balanced, equilibrium, encapsulated by the motor compensation coefficient. This measure assesses the brain’s ability to maintain motor function despite dopamine deficits. It includes an asymmetry score for motor and dopaminergic imaging data, paired with a hemispheric ratio that accounts for the mismatch between motor signs and the dopaminergic deficit. It emerges as a potential biomarker of individual, and hemisphere-specific, compensation for dopaminergic terminal loss, as it is associated with motor cortex activity, connectivity and plasticity.
This perspective could reshape our understanding of Parkinson’s disease, especially within the context of the new biologically defined staging system proposed by Simuni and colleagues.7 In this framework, individuals with a positive seeding assay for α-synuclein (i.e. harbouring abnormal protein accumulation) and/or with striatal dopaminergic denervation can be classified as having biologically defined synucleinopathy. This staging acknowledges that individuals, despite showing molecular and neurodegenerative changes, may remain symptom-free for the rest of their lives. This is exemplified by people who exhibit severe nigrostriatal degeneration without ever experiencing clinical signs of parkinsonism,8 as well as by individuals with REM sleep behaviour disorder (RBD) who do not progress to clinically overt synucleinopathy. Clinicopathological studies are needed to investigate whether these compensatory changes also characterize individuals with incidental Lewy body disease at post-mortem. Overall, it is obvious that key pieces of the puzzle are missing, including an understanding of what drives nigrostriatal degeneration and, more critically, what leads from degeneration to the onset of motor symptoms. Here, Passaretti and colleagues5 show that compensatory mechanisms may fully or partially explain this disconnect, decoupling biological and pathological changes from clinical manifestations.
In this context, compensatory mechanisms should not be considered merely a footnote in the progression of Parkinson’s disease. They are central to how we define and approach the disorder. If compensatory mechanisms are effective enough not only to delay but also to prevent the onset of Parkinson’s disease, they could serve as a blueprint for future therapies. By recognizing and studying these adaptations, we can go beyond merely addressing symptoms, and can instead work to enhance the brain’s inherent ability to cope with, and even overcome, neurodegeneration. Future research should focus on motor compensation across different disease stages, particularly in preclinical or prodromal states. It should investigate the biological and neurophysiological determinants of compensation and seek to identify the point at which the brain can no longer adapt to dopaminergic loss, leading to the failure of compensation. Such efforts will contribute to the transition toward personalized interventions for Parkinson’s disease.
In conclusion, this study offers methodological solutions and invaluable insights into the brain’s adaptive capabilities, challenging our approach to neurodegenerative disorders and shifting our focus towards enhancing the brain’s natural defences: its compensatory mechanisms. As research advances, these findings could pave the way for more personalized and effective therapies, tailored to how each brain uniquely responds to the challenges of neurodegeneration.
Funding
No funding was received towards this work.
Competing interests
The authors report no competing interests.


{kind=link}