




Abstract
Alzheimer’s disease, the most common cause of dementia, is a chronic degenerative disease with typical pathological features of extracellular senile plaques and intracellular neurofibrillary tangles and a significant decrease in the density of neuronal dendritic spines. Cdc42 is a member of the small G protein family that plays an important role in regulating synaptic plasticity and is regulated by Cdc42GAP, which switches Cdc42 from active GTP-bound to inactive GDP-bound states regulating downstream pathways via effector proteins. However, few studies have focused on Cdc42 in the progression of Alzheimer’s disease.
In a heterozygous Cdc42GAP mouse model that exhibited elevated Cdc42-GTPase activity accompanied by increased Cdc42-PAK1-cofilin signalling, we found impairments in cognitive behaviours, neuron senescence, synaptic loss with depolymerization of F-actin and the pathological phenotypes of Alzheimer’s disease, including phosphorylated tau (p-T231, AT8), along with increased soluble and insoluble Aβ1–42 and Aβ1–40, which are consistent with typical Alzheimer’s disease mice. Interestingly, these impairments increased significantly with age. Furthermore, the results of quantitative phosphoproteomic analysis of the hippocampus of 11-month-old GAP mice suggested that Cdc42GAP deficiency induces and accelerates Alzheimer’s disease-like phenotypes through activation of GSK-3β by dephosphorylation at Ser9, Ser389 and/or phosphorylation at Tyr216. In addition, overexpression of dominant-negative Cdc42 in the primary hippocampal and cortical neurons of heterozygous Cdc42GAP mice reversed synaptic loss and tau hyperphosphorylation. Importantly, the Cdc42 signalling pathway, Aβ1–42, Aβ1–40 and GSK-3β activity were increased in the cortical sections of Alzheimer’s disease patients compared with those in healthy controls.
Together, these data indicated that Cdc42GAP is involved in regulating Alzheimer’s disease-like phenotypes such as cognitive deficits, dendritic spine loss, phosphorylated tau (p-T231, AT8) and increased soluble and insoluble Aβ1–42 and Aβ1–40, possibly through the activation of GSK-3β, and these impairments increased significantly with age. Thus, we provide the first evidence that Cdc42 is involved in the progression of Alzheimer’s disease-like phenotypes, which may provide new targets for Alzheimer’s disease treatment.
Introduction
Alzheimer’s disease (AD) is characterized by massive neurodegeneration and altered neuronal connectivity and is histopathologically defined by extracellular amyloid plaques consisting of aggregated amyloid-β (Aβ) and intracellular neurofibrillary tangles (NFTs).1-3 NFTs are composed of hyperphosphorylated forms of the microtubule-associated protein tau, whereas Aβ is derived from the proteolytic cleavage of Aβ precursor protein (APP).
Dendritic spines are highly dynamic structures that are strongly related to synaptic plasticity, learning and memory.4 Filamentous actin (F-actin) is the major cytoskeletal protein in spines and can be depolymerized to monomeric globular actin (G-actin). Polymerization and depolymerization of actin (F/G-actin) is crucial in regulating spine morphology.5 In patients with AD, both amyloid plaques and NFTs lead to alteration and progressive loss of synaptic efficacy accompanied by decreased synaptosomal F-actin levels.6 Synaptic loss is a major contributor to cognitive impairment in AD and has been studied extensively in recent years.7-11
The Rho GTPase family of small G proteins is an important mediator of actin dynamics and plays a critical role in regulating spine morphological changes. For example, Rac1 and RhoA, two members of the Rho GTPase family, have been implicated in synaptic loss in AD.12-16 Cdc42 is another member of the Rho GTPase family and functions similarly to Rac1.17 Cdc42 cycles between the inactive form, GDP-bound state, and the active form, GTP-bound state, in cells and is controlled by many regulators. Cdc42 activation promotes dendritic growth and spine formation, and conditional Cdc42 deletion induces spine loss.18 Cdc42 GTPase-activating protein (Cdc42GAP) is a negative regulator of Cdc42, leading to Cdc42 inactivation. Previous studies have shown that Cdc42GAP knockout homozygous mouse models exhibit premature phenotypes, including reduced body mass and a loss of subdermal adipose tissue.19 Circulating Cdc42 levels are increased in patients with AD, and Cdc42EP4 (Cdc42 effector protein 4) is increased in cortical brain sections.20,21 However, the effects of Cdc42GAP deletion on AD are largely unknown.
The goal of the present study was to investigate whether the activation of Cdc42 signalling is involved in the AD-like phenotype. Using Cdc42GAP knockdown heterozygous mice (GAP mice) and AD mice as a positive control, we found that elevated Cdc42 activity induced and accelerated spine loss with depolymerization of F-actin, neuron senescence and increased tau hyperphosphorylation and Aβ production, which are consistent with typical AD mice, and these impairments increased with age. To further explore the mechanism, we analysed the quantitative phosphoproteomics of the hippocampus of 11-month-old GAP mice, and the results suggested that elevated activity of GSK-3β may result in these impairments. Furthermore, synaptic loss and tau hyperphosphorylation were rescued by overexpression of dominant-negative Cdc42 in the primary hippocampal and cortical neurons of GAP, AD and GAP/AD mice. Importantly, Cdc42 signalling, Aβ1–42, Aβ1–40 and the activity of GSK-3β were increased in cortical sections of AD patients compared with healthy controls. In conclusion, our results indicate that Cdc42 signalling plays an important role in mediating the progression of the AD-like phenotype.
Materials and methods
Animals
We used wild-type mice, GAP heterozygous mice (GAP mice),22 APP/PS1 mice (AD mice) and GAP/AD mice in this study. All procedures were performed in accordance with the National Institutes of Health Guide and approved by the Animal Care and Use Committee of Southern Medical University. The details are fully described in the Supplementary material.
Behavioural assays
Details on behavioural assays are described in the Supplementary material.
Senescence-associated-β-galactosidase staining, immunofluorescence and image acquisition
Brain sections were collected from mice in all groups for senescence-associated-β-galactosidase (SA-β-gal) staining. Details are described in the Supplementary material.
Single-cell microinjection of Lucifer yellow and image acquisition
After the behavioural test, 250-μm coronal sections of brain were collected for single-cell microinjection of Lucifer yellow fluorescent dye. More details can be found in the Supplementary material and have been described previously.23
Cdc42 GTPase pull-down and western blotting
Cdc42/Rac1 GTPase pull-down assays and western blotting were performed as previously described.24 More details are described in the Supplementary material.
Primary cell culture, immunofluorescence staining and image collection
Primary neuronal cultures from wild-type, APP/PS1, Cdc42GAP+/− and GAP/AD mice at postnatal Days 1–3 were obtained and cultured as previously described.25-27 More details are described in the Supplementary material.
Measuring Aβ levels using enzyme-linked immunosorbent assay
Soluble and insoluble Aβ1–40 and Aβ1–42 were measured by enzyme-linked immunosorbent assay (ELISA). More details are described in the Supplementary material.
Assay of F-actin and G-actin
F-actin and G-actin fractions were separated using the G-actin/F-actin In Vivo Assay Kit according to the manufacturer’s instructions (Cytoskeleton, BK037). The samples were quantitated by SDS-PAGE and western blot analysis. More details are described in the Supplementary material.
Quantitative phosphoproteomic analysis
Phosphoproteomic analysis was performed at the Jingjie PTM BioLab Co. Ltd. Proteomics Facility. More details are described in the Supplementary material.
Lentivirus
Lentiviruses containing Cdc42-CA (constitutively active Cdc42) (Cdc42L61) and Cdc42-DN (dominant-negative Cdc42) (Cdc42N17) driven by the cytomegalovirus (CMV) promoter were combined with EGFP and constructed by OBiO. The control lentivirus expressed EGFP alone.
Human subjects
Paraffin-embedded blocks of human post-mortem cortical tissue sections from patients with AD and healthy control subjects were acquired from Guizhou Medical University, and further information is shown in Supplementary Table 1. The subjects’ consent was obtained according to the Declaration of Helsinki and approved by the Medical Ethics Committee of Southern Medical University.
Immunohistochemistry staining
We detected the level of p-PAK1 (Ser199), p-cofilin (Ser3), Cdc42 GTPase, total-Cdc42, C-terminal fragments (CTFs), Aβ1–42, Aβ1–40, p-GSK-3β (Ser9), p-GSK-3β (Tyr216) and total GSK-3β in the cortical tissue sections of AD patients and healthy control subjects by immunohistochemistry. More details are described in the Supplementary material.
Statistical analysis
All statistical analyses were performed using SPSS 22.0. One-way ANOVA was used for comparisons across multiple groups; Bonferroni’s test was used for post hoc comparison. An independent Student’s t-test was performed for comparisons between two groups. P < 0.05 was considered statistically significant. The variance was similar between the compared groups in their respective experiments. All values represent the mean ± standard error of the mean (SEM). All analyses and quantifications were performed in a double-blinded fashion.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary material. The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Results
Cdc42GAP deficiency impaired cognitive and spatial memory
Previous research has demonstrated that Cdc42GAP knockout homozygous mice exhibit premature phenotypes such as body mass loss, senescence in multiple tissues and chromosomal instability.19 However, the role of Cdc42GAP deficiency in AD remains unknown. Thus, in addition to evaluating Cdc42GAP knockout heterozygous mice (GAP mice), we employed APP/PS1 mice (AD mice), a typical mouse model of AD, as a positive control to determine the degree of memory impairment.
We collected the hippocampal and cortical tissues of 11-month-old wild-type, GAP and AD mice to detect Cdc42 signalling and found that active but not total Cdc42 was significantly increased in the hippocampus and cortex of GAP and AD mice (Supplementary Fig. 1A–D). The downstream targets of Cdc42 signalling, including the phosphorylation levels of PAK and cofilin, were all increased (Supplementary Fig. 1A–D), demonstrating that the Cdc42 signalling pathway was elevated in Cdc42GAP-deficient and AD mice. In addition, we crossed GAP mice with AD mice to create GAP/AD mice, which have the phenotype of AD mice with Cdc42GAP deficits. Furthermore, we used mice at 4 and 11 months of age, representing the young and aged stages, to explore the influence of Cdc42GAP deficiency on the process of cognitive degeneration.
We conducted several behavioural tests at different ages to investigate the role of Cdc42GAP deficiency in memory and recognition (Fig. 1A). In the novel object recognition test, we found that in 4-month-old mice, GAP mice spent less time interacting with new objects and interacted with new objects less frequently than wild-type mice (Fig. 1B and C). Furthermore, GAP/AD mice performed worse than GAP mice (Fig. 1B and C). In addition, among the 11-month-old mice, AD mice (the positive control) exhibited worse recognitive ability than wild-type mice. GAP mice performed similarly to AD mice. Moreover, GAP/AD mice explored the novel object less frequently and for a shorter period than AD mice. Interestingly, 11-month-old GAP mice exhibited more severe recognition and memory impairment than younger mice. These results indicate that Cdc42GAP deficiency induces cognitive impairment, which may worsen with age.
![Cdc42GAP deficiency induces cognitive abnormalities, and, with age, more severe behavioural deficits arise. (A) Behavioural test workflow. (B and C) Four-month-old GAP mice (n = 19) spent less time locating the new object and interacted less frequently with the new object than wild-type (WT) mice (n = 26), and Cdc42GAP deficiency induced significantly reduced seeking and interaction with the new object in GAP/AD mice (n = 6). Eleven-month-old GAP mice behaved similarly to those at 4-months of age across groups; cognitive impairment was exacerbated with age [wild-type (n = 24), GAP (n = 40), AD (n = 12) and GAP/AD (n = 7)]. (D and E) The number of shutters in 4- and 11-month-old GAP mice (n = 19) was significantly higher than that in wild-type mice (n = 26), and Cdc42GAP deficiency induced a significantly higher number of shutters in GAP/AD mice (n = 6). While the latency of GAP mice was aberrantly lower than that of wild-type mice, Cdc42GAP deficiency in AD mice led to the lowest latency among the groups, and more severe memory impairment was exhibited in GAP mice with age [wild-type (n = 24), GAP (n = 40), AD (n = 12) and GAP/AD (n = 7)]. (F and G) The total distance required to find the hidden platform and the number of platform crossings in 4-month-old wild-type (n = 13), GAP (n = 12) and GAP/AD (n = 8) mice and 11-month-old wild-type (n = 24), GAP (n = 31) and AD (n = 12) mice were not significantly different. (H and I) At the age of 4 months, the number of platform crossings by GAP mice was significantly lower than that by wild-type mice. At the age of 11 months, significantly fewer platform crossings were observed in GAP and AD mice than in wild-type mice, and older GAP mice exhibited fewer platform crossings than younger mice. Almost the same performance was observed in the reverse probe trial as in the probe trial. One-way ANOVA was used across groups, and the Bonferroni test was employed for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01. NOR = novel novel object recognition test; PAT = passive avoidance test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/146/10/10.1093_brain_awad184/1/m_awad184f1.jpeg?Expires=1750331637&Signature=M3eVb9UDOD-9Vkb7oFvneggNYUNE3LZYqUWkUwXHg7MVeeD3UzMjJg4-sK3mOGhAkHrLSIETz59cv9US2QRDFFGtn9TZiakL6CTNdUgGekOZ55wdESaIlJY92YQr0WNxzrroQ9SFB4kNhrRzNV3BNYOlNfgDeBHHco0V5gSOL8C~Y1XzrUlPgkJoN8YgthtfItWV6zHtRgpkiu2Cx8Buy6U0Afebo2ldjWCyHuNJjpMBuY5C49lrVPniTejFaZRHnCwbiMtmITHI9v-nSPC-SE5rKYA8oum54lz5bmYflngmO1hLpSsUNryw7UVSYqXFd5qtSyoNXeXPDWk61q5bTg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cdc42GAP deficiency induces cognitive abnormalities, and, with age, more severe behavioural deficits arise. (A) Behavioural test workflow. (B and C) Four-month-old GAP mice (n = 19) spent less time locating the new object and interacted less frequently with the new object than wild-type (WT) mice (n = 26), and Cdc42GAP deficiency induced significantly reduced seeking and interaction with the new object in GAP/AD mice (n = 6). Eleven-month-old GAP mice behaved similarly to those at 4-months of age across groups; cognitive impairment was exacerbated with age [wild-type (n = 24), GAP (n = 40), AD (n = 12) and GAP/AD (n = 7)]. (D and E) The number of shutters in 4- and 11-month-old GAP mice (n = 19) was significantly higher than that in wild-type mice (n = 26), and Cdc42GAP deficiency induced a significantly higher number of shutters in GAP/AD mice (n = 6). While the latency of GAP mice was aberrantly lower than that of wild-type mice, Cdc42GAP deficiency in AD mice led to the lowest latency among the groups, and more severe memory impairment was exhibited in GAP mice with age [wild-type (n = 24), GAP (n = 40), AD (n = 12) and GAP/AD (n = 7)]. (F and G) The total distance required to find the hidden platform and the number of platform crossings in 4-month-old wild-type (n = 13), GAP (n = 12) and GAP/AD (n = 8) mice and 11-month-old wild-type (n = 24), GAP (n = 31) and AD (n = 12) mice were not significantly different. (H and I) At the age of 4 months, the number of platform crossings by GAP mice was significantly lower than that by wild-type mice. At the age of 11 months, significantly fewer platform crossings were observed in GAP and AD mice than in wild-type mice, and older GAP mice exhibited fewer platform crossings than younger mice. Almost the same performance was observed in the reverse probe trial as in the probe trial. One-way ANOVA was used across groups, and the Bonferroni test was employed for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01. NOR = novel novel object recognition test; PAT = passive avoidance test.
In the passive avoidance test, we found that in 4-month-old mice, latency was significantly shorter, and the number of shutters was increased in GAP mice compared with wild-type mice. GAP/AD mice had similar results to GAP mice. In addition, among the 11-month-old mice, the latency time of AD and GAP mice was significantly shorter than that of wild-type mice. There were no differences among GAP, AD and GAP/AD mice (Fig. 1D). Similarly, the number of shutters was significantly elevated in GAP, AD and GAP/AD mice compared with wild-type mice (Fig. 1E). In particular, compared to 4-month-old GAP mice, 11-month-old GAP mice had a shorter latency time (Fig. 1D). These findings suggest that Cdc42GAP deficiency is also related to recognition and memory impairment.
The Morris water maze test was used to investigate the effect of Cdc42GAP deficiency on spatial memory. First, there was no difference in athletic ability or performance after brief training of all mice in this experiment (Fig. 1F and G). In 4-month-old mice, GAP and GAP/AD mice all showed decreased platform crossings during probe and reverse probe trials compared with wild-type mice. There was no difference between GAP and GAP/AD mice. Similarly, 11-month-old GAP and AD mice performed worse than wild-type mice, and older GAP mice performed worse than younger GAP mice (Fig. 1H and I). Based on the findings of this study, Cdc42GAP deficiency impairs spatial memory.
Cdc42GAP deficiency induced neuron senescence and spine degeneration with depolymerization of F-actin
Previous research has indicated that increased Cdc42 activity can cause cell apoptosis in various tissues.28,29 The hippocampus is an important mediator of cognition and memory, as it receives and integrates information from the cortex and then connects to other regions. We detected SA-β-gal, a neuron senescence marker, in hippocampal and cortical neurons to evaluate Cdc42GAP deficiency in neuronal ageing. We found that the SA-β-gal level in 4-month-old GAP mice was significantly increased compared with that in wild-type mice. In addition, 4-month-old GAP/AD mice exhibited more severe senescence than GAP mice. Furthermore, neuron senescence was increased in 11-month-old mice. GAP and AD mice exhibited elevated SA-β-gal expression compared with wild-type mice. We also found that the degree of ageing in 11-month-old GAP/AD mice was significantly more serious than that in GAP and AD mice, indicating that Cdc42GAP deficiency induced and accelerated neuronal senescence (Fig. 2).
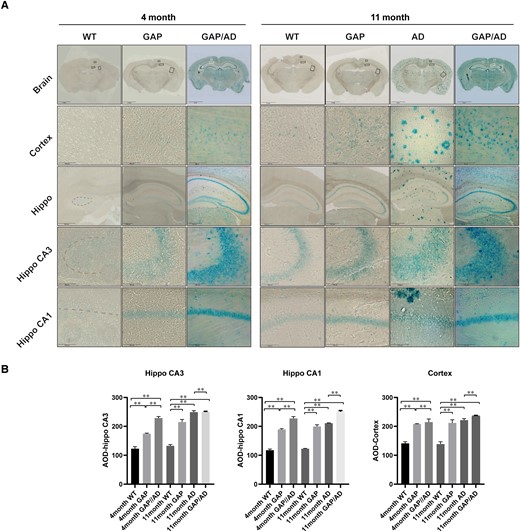
Cdc42GAP deficiency induces neuron senescence. (A) Senescence-associated β-gal staining in GAP, AD and GAP/AD mice was higher than that in wild-type mice in the hippocampus and cortex, and Cdc42GAP deficiency may worsen neuronal senescence in AD mice. Meanwhile, 11-month-old GAP mice exhibited a higher signal than 4-month-old GAP mice. (B) Quantification of the average optical density in A. One-way ANOVA was used across groups, and the Bonferroni test was employed for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01.
In addition, spine density in visual cortex pyramidal and hippocampal neurons is strongly related to brain function.30,31 Thus, we investigated the role of Cdc42GAP in spine plasticity in these two brain regions. After behavioural testing, we prepared brain slices and patch-clamped neurons in 250-µm vibrating slices (Fig. 3A and B). The following three distinct subtypes of spines in neurons were identified: thin spines, stubby spines and mushroom spines. In the hippocampal CA3 region, the total spine and three distinct spine densities of GAP mice were decreased significantly compared to those of wild-type mice at 4 and 11 months. Additionally, in 4-month-old mice, the total spine, mushroom spine and thin spine densities of GAP/AD mice were decreased compared with those of GAP mice. In 11-month-old mice, GAP mice exhibited fewer total and distinct spine densities than AD mice, while GAP/AD mice were approximately the same as GAP mice. Specifically, total spine density in the older GAP mice, particularly of mushroom and stubby spines, was significantly reduced compared to that of younger mice (Fig. 3D). Meanwhile, there were similar values in the hippocampal CA1 region (Fig. 3E) and cortex (Fig. 3F). The substantial reduction in total spine density was primarily due to a decline in mushroom and stubby spines. These findings demonstrated that Cdc42GAP deficiency induces long-term disruptions in rodent synaptic plasticity in the hippocampus and cortex, which are associated with cognition and memory.
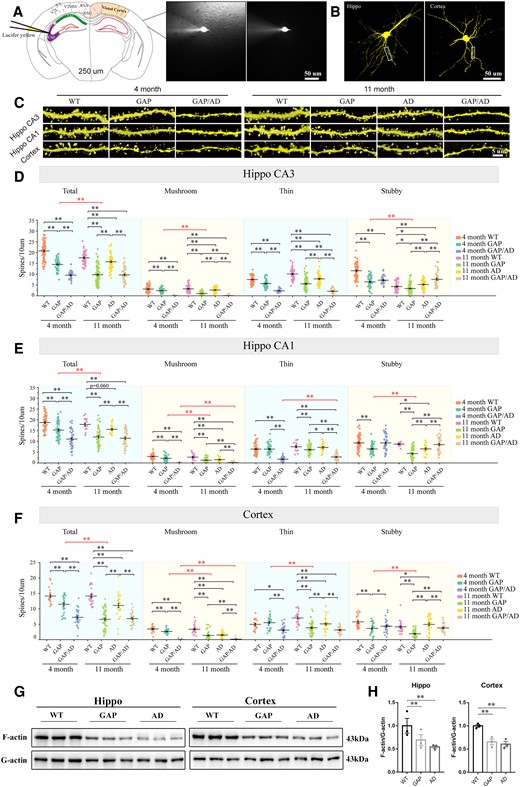
Cdc42GAP induces spine remodelling. (A and B) Illustration of the microinjection of patch-clamped neurons and 3D reconstruction of hippocampal and cortical neurons. (C) Representative images of dendritic spines of mice of different genotypes at younger and older ages. (D) At the age of 11 months, quantification of total, mushroom and stubby spines in the hippocampal CA3 region revealed significantly decreased spine counts in GAP mice compared with wild-type (WT) and AD mice, and Cdc42GAP deficiency may worsen spine loss in AD mice. Similar findings were found in 4-month-old mice, and the total spine density was lower in 11-month-old than 4-month-old GAP mice. (E) At the age of 11 months, quantification of total, mushroom and stubby spines in the hippocampal CA1 region showed significantly decreased spine counts in GAP mice compared with wild-type and AD mice, and Cdc42GAP deficiency may worsen spine loss in AD mice. Similar findings were found in 4-month-old mice, and the total spine density was lower in 11-month-old than 4-month-old GAP mice. (F) At the age of 11 months, quantification of total, mushroom and stubby spines in the cortex demonstrated significantly decreased spine counts in GAP mice compared with wild-type and AD mice, and Cdc42GAP deficiency may deteriorate spine loss in AD mice. Similar findings were found in 4-month-old mice, and the total spine density was lower in 11-month GAP mice than in 4-month-old GAP mice. Representative western blots (G) and statistical analyses (H) indicated that the ratio of the levels of F- and G-actin in the hippocampus and cortex of 11-month-old GAP and AD mice was significantly decreased compared to that in wild-type mice (n = 3 mice/group). One-way ANOVA was used across groups, and the Bonferroni test was employed for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01.
Furthermore, we examined F- and G-actin in the hippocampus and cortex of 11-month-old mice. Remarkably, the ratio of F-/G-actin was significantly decreased in the hippocampus and cortex of GAP and AD mice compared with the wild-type mice (Fig. 3G and H), indicating that Cdc42GAP deficiency induces depolymerization of F-actin.
Cdc42GAP deficiency induced and accelerated tau phosphorylation and amyloid-β production
Brain intracellular NFTs, characterized by hyperphosphorylated tau proteins, are a typical phenotype of neuronal impairment in AD. Phosphorylated Thr231 and AT8 (Ser202 and Thr205) are simultaneously two common phosphorylation sites. Furthermore, in the brains of AD patients, the activity of γ-secretase is aberrantly elevated, resulting in several Aβ peptides of varying lengths. Extracellular insoluble amyloid plaques, primarily composed of Aβ1–40 and Aβ1–42, are another neuropathological feature of AD.32,33 Thus, we used 11-month-old mice to investigate the role of Cdc42GAP deficiency in tau phosphorylation and Aβ production.
The immunofluorescence analysis of brain sections revealed that tau phosphorylation (p-T231/AT8) was significantly elevated in the hippocampus and cortex of GAP mice compared to that in wild-type mice (Fig. 4A–D). Simultaneously, tau hyperphosphorylation in GAP/AD mice was further increased compared to that in GAP and AD mice, indicating that Cdc42GAP deficiency may contribute to the progression of tau phosphorylation.
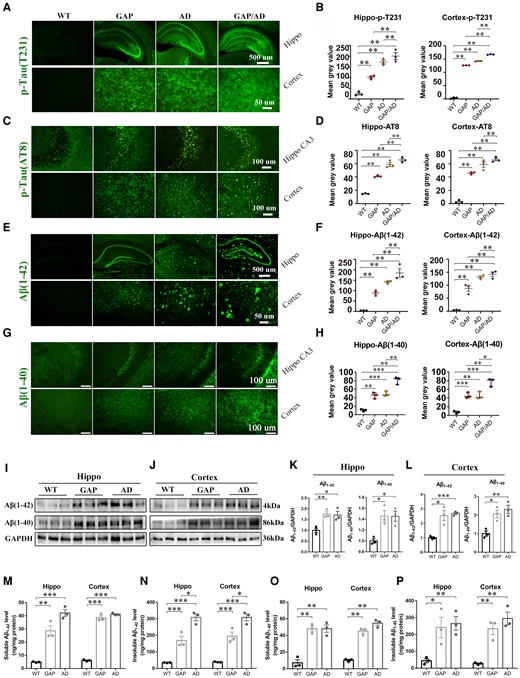
Cdc42GAP deficiency induced AD phenotypes in 11-month-old mice. (A–D) Immunofluorescence images (left) and quantification of the average optical density analyses (right) showed that the levels of phosphorylated Tau (p-T231) (A and B) and Tau (AT8) (C and D) in GAP, AD and GAP/AD mice were greater than those in wild-type (WT) mice in the hippocampus and cortex, and Cdc42GAP deficiency worsened tau phosphorylation in AD mice (n = 3 mice/group). (E–H) Immunofluorescence images (left) and quantification of the average optical density analyses (right) showed that Aβ1–42 (E and F) and Aβ1–40 (G and H) in GAP, AD and GAP/AD mice were significantly increased compared with those in wild-type mice and that Cdc42GAP deficiency worsened Aβ aggregation (n = 3 mice/group). (I–L) Representative western blots (I and J) and statistical analyses (right) revealed that the levels of Aβ1–42 and Aβ1–40 in the hippocampus (K) and cortex (L) from GAP and AD mice were elevated compared with those in wild-type mice (n = 3 mice/group). (M–P) ELISA analyses indicated that the levels of soluble (M) and insoluble (N) Aβ1–42, and soluble (O) and insoluble (P) Aβ1–40, were significantly increased in the hippocampus and cortex of GAP and AD mice compared with wild-type mice (n = 3 mice/group). One-way ANOVA was used across groups, and the least significant difference test was used for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
On the other hand, the immunofluorescence results revealed that Aβ1-42 and Aβ1-40 in the cortex and hippocampus of 11-month-old GAP and AD mice were elevated compared with wild-type mice (Fig. 4E–H). Interestingly, in GAP mice, Aβ1–42 was concentrated in the neurons instead of senile plaques accumulating outside of the cell membrane. In addition, Aβ1–42 and Aβ1–40 levels in GAP/AD mice were significantly increased compared to those in GAP and AD mice (Fig. 4E–H). Additionally, we collected the hippocampal and cortical tissues of 11-month-old mice. Using western blotting, we also found that Aβ1–42 and Aβ1–40 in the cortex and hippocampus in GAP and AD mice were increased compared with those in wild-type mice (Fig. 4I–L). On the other hand, we analysed soluble and insoluble Aβ1–42 and Aβ1–40 in 11-month-old mice by ELISA. We observed that the levels of soluble Aβ1–42 in the cortex and hippocampus of GAP and AD mice were higher than in wild-type mice (Fig. 4M). Insoluble Aβ1–42 in the cortex and hippocampus of GAP and AD mice was also elevated compared with wild-type mice. Interestingly, AD mice had more insoluble Aβ1–42 than GAP mice (Fig. 4N). These results indicated that Cdc42GAP deficiency induced the metabolic disturbance of insoluble Aβ1–42 but did not change the accumulation of insoluble Aβ1–42. Similar to Aβ1–42, in GAP and AD mice, the levels of soluble and insoluble Aβ1–40 in the cortex and hippocampus were significantly elevated compared with wild-type mice (Fig. 4O and P). There was no difference between GAP and AD mice.
In addition, we analysed the levels of sAPPα and APP-CTFβ, which are the products of two APP processing pathways. The immunofluorescence analysis of brain sections revealed that CTFs, which are produced by APP in amyloidogenic processing, were increased in the hippocampus and cortex of 11-month-old GAP, AD and GAP/AD mice. Specifically, CTFs in GAP/AD mice were significantly elevated compared with GAP and AD mice (Supplementary Fig. 2A and B). Western blotting revealed the same results (Supplementary Fig. 2C and D). In contrast, the level of sAPPα, which is produced by APP in nonamyloidogenic processing, was significantly reduced in the hippocampus and cortex of 11-month-old GAP and AD mice compared with wild-type mice (Supplementary Fig. 2E). Collectively, these results suggested that Cdc42GAP deficiency induces and accelerates tau phosphorylation and production of Aβ1–40 and Aβ1–42.
Cdc42GAP deficiency induces Alzheimer’s disease phenotype, probably by activating GSK-3β
To explore the potential mechanism of the AD phenotype in Cdc42GAP-deficient mice, we conducted a quantitative phosphoproteomic analysis of the hippocampus of 11-month-old wild-type and GAP mice (n = 3). Compared to wild-type mice, we found that 109 phosphoproteins and 134 sites were significantly upregulated, and 154 phosphoproteins and 202 sites were downregulated in GAP mice. The magnitude and significance of changes in the protein phosphorylation level between wild-type and GAP mice are presented in a volcano plot (Fig. 5A). Moreover, the results of a gene ontology enrichment analysis showed that differentially expressed phosphorylated proteins participated in negative regulation of cytoskeleton and amyloid fibril formation (Fig. 5B). Among the upregulated phosphoproteins, microtubule-associated protein tau (MAPT) was phosphorylated at many sites, which is consistent with the immunofluorescence results in that the level of tau phosphorylation was significantly elevated in the hippocampus and cortex of GAP mice. In addition, we found that phosphorylation of glycogen synthase kinase-3β (GSK-3β) at Ser389 and Ser215 was downregulated (Fig. 5A). Numerous studies have shown that dephosphorylation of GSK-3β at Ser9 and Ser389 or phosphorylation at Tyr216 can activate GSK-3β and that overactivated GSK-3β is involved in the progression of AD.34-36 Thus, we analysed the activity of GSK-3β at Ser389, Ser9 and Tyr216 by western blotting and immunofluorescence. Compared with 11-month-old wild-type mice, the levels of phosphorylation of GSK-3β at Ser389 and Ser9 were both significantly decreased in the hippocampus and cortex of GAP and AD mice (Fig. 5C–F). In contrast, total GSK-3β and the level of phosphorylation of GSK-3β at Tyr216 were significantly increased in GAP and AD mice (Fig. 5C–F). The immunofluorescence results further showed that the level of phosphorylated GSK-3β Ser9 was reduced in the hippocampus and cortex of 11-month-old GAP, AD and GAP/AD mice compared with wild-type mice (Fig. 5G and H). Notably, the level of phosphorylated GSK-3β Ser9 in the hippocampus of GAP/AD mice was lower than that in AD mice, and phosphorylated GSK-3β Ser9 in the cortex of GAP/AD mice was also lower than that in AD as well as GAP mice (Fig. 5G and H). On the other hand, the immunofluorescence results revealed that the level of phosphorylated GSK-3β Tyr216 was increased in the hippocampus and cortex of 11-month-old GAP, AD and GAP/AD mice compared with wild-type mice, with the level of Tyr216 in the cortex of GAP/AD mice being higher than that in GAP and AD mice (Fig. 5I and J).
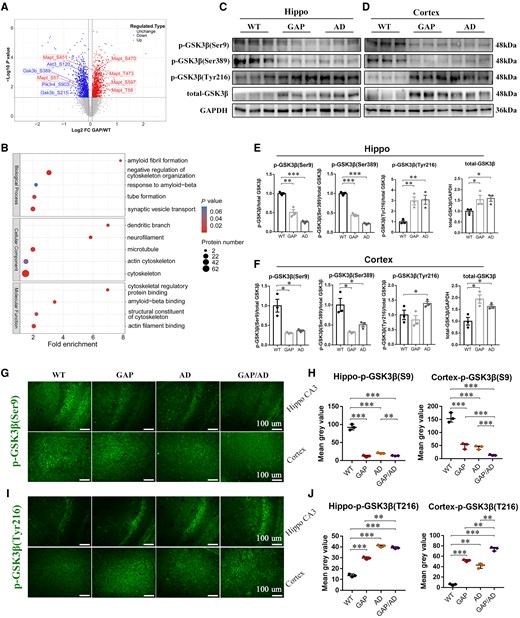
Cdc42GAP deficiency may induce AD phenotypes by activating GSK-3β. (A) Volcano plot showing the significantly changed proteins between 11-month-old wild-type (WT) and GAP mice (P < 0.05). Blue dots represent downregulated phosphorylated proteins, and red dots represent upregulated phosphorylated proteins. (B) Bubble chart showing the gene ontology enrichment analysis of the differentially expressed proteins upregulated in GAP mice compared to wild-type mice. The bubble size represents the number of differentially expressed proteins, and the bubble colour represents the P-value. (C–F) Representative western blots (C and D) and statistical analyses suggested that the activity of GSK-3β and total GSK-3β were significantly elevated in the hippocampus (E) and cortex (F) of GAP and AD mice compared with wild-type mice (n = 3 mice/group). (G and H) Immunofluorescence images (left) and quantification of the average optical density analyses (right) showed that the level of phosphorylated GSK-3β at Ser9 was reduced in the hippocampal CA3 region and cortex of GAP, AD and GAP/AD mice compared with wild-type mice (n = 3 mice/group). (I and J) Immunofluorescence images (left) and quantification of the average optical density analyses (right) showed that the level of phosphorylated GSK-3β at Tyr216 was increased in the hippocampal CA3 region and cortex of GAP, AD and GAP/AD mice compared with wild-type mice (n = 3 mice/group). One-way ANOVA was used across groups, and the least significant difference test was used for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
In general, these findings imply that Cdc42GAP deficiency induces and accelerates AD-like phenotypes in the hippocampus and cortex through activating GSK-3β by dephosphorylation at Ser9, Ser389 and/or phosphorylation at Tyr216.
Persistent activation of Cdc42 GTPase induced tau hyperphosphorylation and synaptic loss
We first investigated Cdc42, a small GTPase with an established role in actin remodelling, is a key regulator of AD phenotypes. Sequencing-based studies have suggested that Cdc42 changes in the AD process,21,37 and we manipulated Cdc42 activity to verify the mechanism of Cdc42GAP deficiency in tau hyperphosphorylation and spine loss.
We obtained and cultured primary hippocampal and cortical samples from wild-type, GAP, AD and GAP/AD mice at postnatal Days 1–3 and then treated them with Cdc42-related lentiviruses (Fig. 6A). Our previous publications confirmed that Cdc42-CA efficiently increased Cdc42 activity and its downstream signalling activities, and Cdc42-DN decreased Cdc42 activity and its downstream signalling activities.18,23 All groups received a GFP lentivirus, and almost no expression of phosphorylated tau was observed in the wild-type groups, whereas overexpression was observed in the GAP, AD and GAP/AD groups. Unlike GFP control lentivirus treatment of primary neuronal cultures, treatment with 0.5 µl (1 × 108) Cdc42-DN lentivirus for 2 weeks (Fig. 6A) significantly decreased tau phosphorylation in the GAP, AD and GAP/AD groups (Fig. 6B–E). In contrast, when treated with Cdc42-CA lentivirus, tau phosphorylation in the GAP, AD and GAP/AD groups was increased compared to that in the GFP control lentivirus-treated groups (Fig. 6B–E). These results illustrate that elevated Cdc42 GTPase activity is responsible for AD-like disruptions in tau hyperphosphorylation.
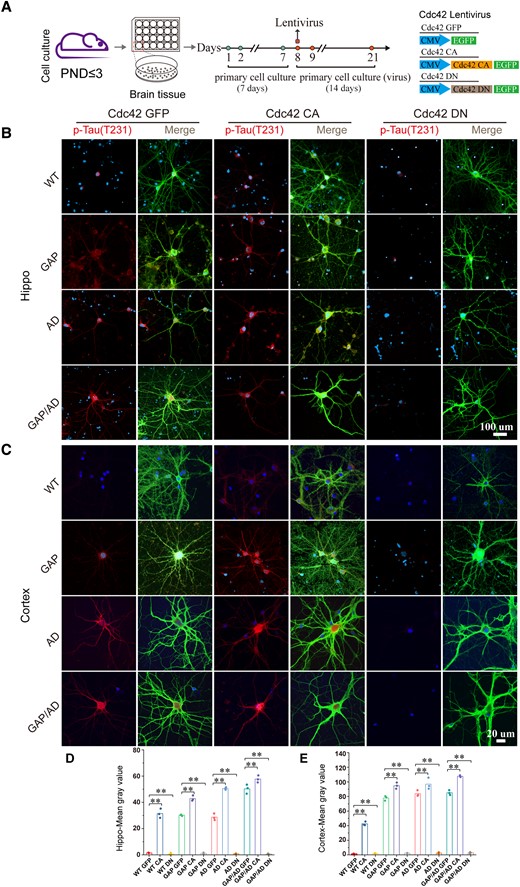
Elevated Cdc42 GTPase activity increased phosphorylated tau levels. (A) Flow chart of cell culture. (B and C) Immunofluorescence staining of tau phosphorylation (p-T231) in live-cultured fresh hippocampal and cortical neurons of wild-type, GAP, AD and GAP/AD mice showed that elevated Cdc42 GTPase activity could induce tau hyperphosphorylation and downregulate tau phosphorylation levels when Cdc42-DN lentivirus was used. (D and E) Quantification of the mean grey values in B and C. One-way ANOVA was used across groups; for post hoc comparison, the least significant difference test was used. Error bars depict SEM. *P < 0.05, **P < 0.01. PND = postnatal Day. CA = constitutively active; DN = dominant-negative.
Next, we analysed hippocampal and cortical synaptic plasticity (Fig. 7A and B). After treatment with Cdc42-CA, we found that the total spine density, specifically of mushroom and stubby spines, was decreased in the primary hippocampal neurons of wild-type and GAP mice compared with those that received GFP treatment, and the total spine density of AD and GAP/AD mice was not different. Meanwhile, the total spine density, specifically of mushroom and stubby spines, was significantly decreased in the primary cortical neurons of GAP, AD and GAP/AD mice. In contrast, after treatment with Cdc42-DN, the total spine density, specifically that of thin and stubby spines, of the hippocampal neurons of GAP, AD and GAP/AD mice, was increased compared to that in those that received GFP treatment (Fig. 7C and D). In addition, cortical neurons of GAP and GAP/AD mice were also increased, but the total spine density of AD mice was not increased by Cdc42 DN treatment; a slight decrease in thin spines was also observed. We also found that constitutively elevated Cdc42 levels induced heightened hippocampal and cortical neuronal senescence levels (Supplementary Figs 3 and 4). These results suggest that elevated Cdc42 GTPase activity is involved in AD-like degenerated synaptic plasticity.
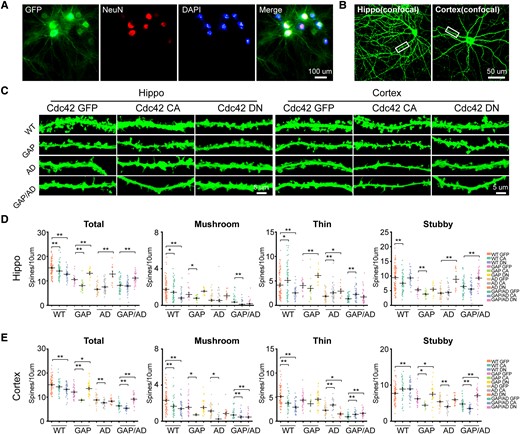
Elevated Cdc42 GTPase activity reduced hippocampal and cortical dendritic spine density. (A and B) Images of cultured hippocampal and cortical neurons. (C) Representative images of dendritic spines of wild-type (WT), GAP, AD and GAP/AD mice in the hippocampus and cortex. (D) The quantification of hippocampal total, mushroom, thin and stubby spines showed significantly decreased spine counts in the Cdc42-CA lentivirus-treated groups, and when treated with Cdc42-DN lentivirus, the reduced synaptic plasticity became closer to normal levels in the GAP, AD and GAP/AD groups. (E) Quantification of cortical total, mushroom, thin and stubby spines showed significantly decreased spine counts in the Cdc42-CA lentivirus-treated groups, and when treated with Cdc42-DN lentivirus, the reduced synaptic plasticity became closer to normal levels in the GAP, AD and GAP/AD groups. One-way ANOVA was used across groups, and the Bonferroni test was used as a post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01. CA = constitutively active; DN = dominant-negative.
Cdc42 signalling pathway and GSK-3β activated in cortical sections of Alzheimer’s disease patients
Despite increased circulating Cdc42 and Cdc42 in the cortex of AD patients,20 the active Cdc42 GTPase levels remain unknown. We obtained cortical paraffin specimens from four patients with AD at Braak stage II or III and four healthy control subjects from Guizhou Medical University and evaluated the Cdc42 signalling pathway and GSK-3β activity by immunochemistry staining. In accordance with the western blotting results of AD mice, there was no difference in total Cdc42, but Cdc42 GTPase in the cortex of AD patients was significantly increased compared with that in the cortex of healthy control subjects (Fig. 8A and B). Furthermore, p-cofilin (Ser3) and p-PAK1 (Ser199) levels in patients with AD were also increased (Fig. 8A and B), indicating that the activated Cdc42 pathway may contribute to AD-associated neuron deterioration in humans. Moreover, Aβ1–40, Aβ1–42 and CTFs, the products of APP processing, were significantly elevated in patients with AD (Fig. 8C and D). We also detected the activity of GSK-3β. The results showed that phosphorylation of GSK-3β at Ser9 was decreased, whereas total GSK-3β and phosphorylation of GSK-3β at Tyr216 were significantly increased (Fig. 8E and F). In general, Cdc42GAP deficiency results in elevated activity of GSK-3β. In conclusion, Cdc42GAP deficiency increases the Cdc42 signalling pathway, and activated GSK-3β plays a vital role in patients with AD.
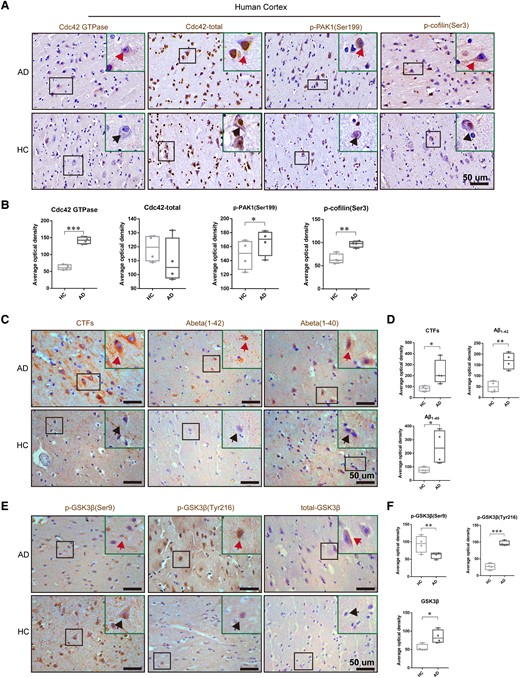
The Cdc42 pathway and GSK-3β activity were activated in the cortex of patients with AD. (A and B) Representative immunohistochemical staining images showed unaltered total Cdc42 and elevated Cdc42 activity, the phosphorylated form of PAK and cofilin in the cortex of AD patients (n = 4). Arrows point to representative cells. (C and D) Representative immunohistochemical staining images showed increased CTFs, Aβ1–42 and Aβ1–40 in the cortex of AD patients. Arrows point to representative cells. (E and F) Representative immunohistochemical staining images showed increased activity of GSK-3β and total GSK-3β. Arrows point to representative cells. Samples were from four patients with AD and four healthy control subjects (HC). One-way ANOVA was used across groups, and the least significant difference test was used for post hoc comparison. Error bars depict SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In this study, we found that Cdc42GAP deficiency induced cognitive impairment and synaptic loss in 4-month-old and 11-month-old mice and worsened with age, along with increased tau phosphorylation and Aβ production, which was consistent with AD mice. In addition, elevated Cdc42 GTPase activity, accompanied by increased Cdc42-PAK1-cofilin signalling, was found in AD mice. Furthermore, the results of the quantitative phosphoproteomic analysis of the hippocampus of 11-month-old GAP mice suggested that elevated activity of GSK-3β may be involved in these impairments. Additionally, the Cdc42 GTPase pathway and GSK-3β activity were activated in patients with AD.
Previous studies have revealed that Cdc42GAP, the negative regulator of Cdc42, significantly shortens the life span of animals and results in multiple other premature ageing-like phenotypes. Numerous studies have demonstrated that Cdc42 activity is related to ageing. For example, the systemic treatment of aged mice with a Cdc42 activity-specific inhibitor significantly extends the average and maximum lifespan.38 Elevated Cdc42 activity in aged haematopoietic stem cells (HSCs) is linked with HSC ageing and related to a loss of polarity in aged HSCs.39 Proteomic analysis of human skin demonstrated that samples from older people have an increased content of Cdc42 compared with younger people.40 In our study, we found that 4-month-old GAP mice showed neuronal ageing at a young age. These impairments worsened with increasing age. In addition, the SA-β-gal activity of GAP/AD mice was significantly increased compared with that of AD mice, indicating that Cdc42GAP deficiency induces and accelerates neuronal ageing. Elevated Cdc42 activity mediates ageing through multiple pathways. For instance, overexpression of constitutively dominant positive Cdc42 in endothelial cells induces H2O2 production, which accelerates the progression of ageing.41 The increased expression of EGFR might be the reason for Cdc42 activation at various tissues during ageing.42,43
In addition to neuron senescence, we observed AD-like cognitive and memory impairment accompanied by spine loss, tau phosphorylation and Aβ production in Cdc42GAP-deficient mice. Aside from Aβ and tau, synapse and dendritic loss show the strongest correlation with cognitive impairment in AD.44 Dendritic spines are key mediators of synaptic transmission, and their structure is linked to synaptic strength and activity. Cdc42, a small GTPase, is a prominent regulator of actin cytoskeleton reorganization. In our study, we found significant decreases in mushroom and stubby spines, which are linked with long-term memory in the hippocampus and cortex of 4-month-old GAP mice, but thin spines, which are related to learning, were not significantly reduced, and these results were consistent in AD patients.22 In addition, we observed more significant decreases in total spines in 11-month-old GAP mice than in AD mice. Given that Cdc42 is a key regulator of dendrite and spine structural plasticity, elevated levels of the active form of Cdc42 in GAP mice may have direct effects on spines. Moreover, we found that after treatment with Cdc42-DN, the total spine density of hippocampal primary neurons in AD mice was rescued to the level seen in wild-type mice; however, the total spine density of cortex primary neurons was not rescued to that found in wild-type mice. Seemingly, the downregulation of Cdc42 activity by Cdc42-DN is not sufficient to rescue spine phenotypes in the cortex of AD mice. However, incomplete rescue is likely due to the decrease (not the increase that was detected in the hippocampus) in the thin spine density of the cortex after treatment with Cdc42-DN. The differential effects of Cdc42-DN on thin spine density in the hippocampus versus the cortex are consistent with previous reports that the plasticity of dendritic spine density in hippocampal and cortical neurons differs in mechanism and time scale, which may be related to their respective functions and circuits. Some studies suggest that dendritic spine density changes can occur more rapidly in the hippocampus, whereas a longer period of time may be required in cortical areas.45,46 Specifically, in the cortex, mushroom and stubby spines, rather than thin spines, are susceptible to chronic stress.47 These differences may reflect the functional demands of different neural systems and their roles in forming and regulating neural circuits.
PAK1 and cofilin are two downstream molecules of Cdc42. The activated Cdc42-PAK1-cofilin signalling pathway in the hippocampus and cortex of 11-month-old GAP and AD mice was observed in the present study. In patients with AD, Cdc42 GTPase was elevated, which has not previously been reported in the cortical tissues of AD patients. A previous study reported that aberrant activation of PAK1 appears around the onset of cognitive deficits in AD.48 We also observed that p-PAK1 mostly aggregates in the nucleus rather than the cytosol in AD patients, which may initiate inflammation-associated transcription.12,45 However, another study has shown that defects in PAK by activating cofilin alter spine dynamics and result in cognitive impairment in AD mice.18 Cofilin, a major actin-depolymerizing factor, plays an essential role in the dynamics of the actin cytoskeleton. The active form of cofilin is regulated by the PAK1-LIM kinase pathway and is upregulated in the cortex of AD patients.49-52 In addition, hyperactivated cofilin might accelerate ageing by inducing brain inflammation and lead to AD.53
Glycogen synthase kinase-3 (GSK3) is a protein-serine/threonine kinase that has two isoforms, glycogen synthase kinase-3 alpha (GSK-3α) and beta (GSK-3β). GSK-3β is mainly expressed in the CNS and activated by dephosphorylation at Ser9 and Ser389 or phosphorylation at Tyr216. Additionally, GSK-3β, as the downstream molecule of Cdc42, is involved in radial glial growth,54 terminal differentiation of hair follicle progenitor cells,55 axonogenesis of primary hippocampal neurons55 and so on. However, it is unclear whether Cdc42 can modulate GSK-3β in AD. In this study, compared to wild-type mice, Cdc42GAP-deficient mice exhibited tau hyperphosphorylation and increased Aβ. Furthermore, we explored the possible mechanism. The quantitative phosphoproteomic analysis of the hippocampus of 11-month-old GAP mice and further verification suggested that phosphorylation of GSK-3β at Ser389 was reduced. In addition, phosphorylation at Ser9 was also decreased and Tyr216 was elevated in GAP, AD mice and AD patients. These results indicated that GSK-3β activity was elevated in Cdc42GAP-deficient mice and patients with AD. It has been reported that a significant increase in the level of GSK-3β (p-Tyr216) is observed in the frontal cortex of patients with AD.56 Numerous studies have shown that increased GSK-3β activity is associated with the progression of AD, such as tau phosphorylation and Aβ production.57,58 GSK-3β phosphorylates at least 36 residues in tau and the main sites are Ser199, Thr-231, Ser396 and Ser413.59 The abnormal upregulation of GSK-3β is strongly associated with the accumulation of toxic tau aggregates.60 Previous studies have demonstrated that GSK-3β is a downstream molecule of the PI3K/Akt signalling pathway. Activated PI3K induces the phosphorylation of Akt at positions 308 and 473, thereby activating Akt, and activated Akt can bind to GSK-3β, phosphorylate it at Ser9 and lead to its inactivation.61 PI3K/Akt/GSK-3β dysfunction results in tau hyperphosphorylation in AD mice.62 In this study, phosphoproteomic analysis showed decreased activity of PI3K and Akt, indicating that PI3K/Akt/GSK-3β dysfunction may mediate tau hyperphosphorylation in Cdc42GAP-deficient mice. However, this hypothesis needs to be further verified. On the other hand, in our study, we found that Aβ1–40 and Aβ1–42 were increased, accompanied by increased CTFs and decreased sAPPα, consistent with AD mice. Overactivated GSK-3β also leads to Aβ production and accumulation. GSK-3β activation is involved in NFK-β/p65 nuclear translocation and promotes the expression of β-secretase (BACE1), which mediates APP amyloidogenic processing.63 Moreover, activated GSK-3β downregulates the efficiency of the PS1/N-cadherin/β-catenin complex which alters PS1/γ-secretase substrate specificity and then increases Aβ production.64 The specific influence of these secretases in Cdc42GAP-deficient mice remains unclear.
In addition, activated GSK-3β is also related to senescence and synaptic plasticity. Both total GSK-3β and activated GSK-3β are increased in the cortex of aged rats.65 In renal diseases, GSK-3β is overexpressed and hyperactive with age in glomerular podocytes66 and inhibited GSK-3β could lessen podocyte senescence.67 As a result, activated GSK-3β may be the potential mechanism of neuronal senescence in Cdc42GAP-deficient mice. On the other hand, the activity of GSK-3β is associated with synaptic plasticity and memory consolidation. Synaptic plasticity is key to the occurrence of long-term potentiation (LTP) and long-term depression (LTD). LTP is defined as an increase in synaptic strength, whereas LTD is the opposite, and the molecular mechanisms involved in these processes are essential to understanding the mechanisms of memory formation. One study indicated that phosphorylation of GSK-3β at Ser9, which inhibited GSK-3β, was increased after inducing LTP in the hippocampus.68 In contrast, elevated GSK-3β activity impaired LTP and promoted LTD.69 In addition, mice expressing constitutively active GSK-3β had longer and thinner dendritic spines, whereas GSK-3β-deficient mice had shorter dendritic spines.70 In general, overactivated GSK-3β impairs cognition and memory by regulating synaptic plasticity.
In conclusion, we found that increased Cdc42 activity caused by Cdc42GAP deficiency induces AD phenotypes possibly by activating GSK-3β, leading to cognitive deficits in AD. Thus, it is tempting to speculate that Cdc42 is a therapeutic target for improving cognitive deficits in AD.
Acknowledgements
We thank Xingmei Zhang (Southern Medical University) for providing AD mice and Rongqing Chen (Southern Medical University) for technical help with patch clamping.
Funding
This work was supported by the Natural Science Foundation of China (81971297, 82073417, 81872514), Guangdong Basic and Applied Basic Research Foundation (2023A1515012480), GDMPA Key Laboratory Project of Scientific and Technological Innovation (2023ZDZ12) and Colleges Pearl River Scholar Funded Scheme (GDUPS2015).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
References
Author notes
Mengjuan Zhu and Bin Xiao contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}