Sir,
We wish to address the recent proposal of a disease entity newly titled ‘limbic-predominant age-related TDP-43 encephalopathy (LATE)’ (Nelson et al., 2019) which, to our reading, is itself a derivative of the 2016 proposed term cerebral age-related TDP-43 with sclerosis (CART) (Nelson et al., 2016). The transactive response DNA binding protein of ∼43 kD (TDP-43) was first reported in 2006 to be a main component of ubiquitinated inclusions in autopsy-confirmed cases of frontotemporal lobar degeneration (FTLD) negative for tau immunoreactivity (Arai et al., 2006; Neumann et al., 2006). Over a decade of research into FTLD-TDP (and FTLD-U before it) has highlighted important phenotypic variability in TDP-43-immunoreactive lesions resulting in the identification of five different types of FTLD-TDP (Mackenzie et al., 2006, 2011; Sampathu et al., 2006; Josephs et al., 2009; Lee et al., 2011, 2017). There have also been important advances in the molecular and biochemical characterization of TDP-immunoreactive in FTLD-TDP (Sampathu et al., 2006; Hasegawa et al., 2008; Igaz et al., 2008; Zhang et al., 2009; Bigio et al., 2013; Laferriere et al., 2019). Soon after the initial characterization of TDP-immunoreactive lesions in FTLD and amyotrophic lateral sclerosis, TDP-immunoreactive lesions were identified in 25–33% of cases with pathologically confirmed Alzheimer’s disease (Amador-Ortiz et al., 2007; Higashi et al., 2007). TDP-immunoreactive lesions related to Alzheimer’s disease were initially thought to appear first in the hippocampus (Amador-Ortiz et al., 2007), but more detailed morphological studies involving multiple anatomical regions revealed the amygdala to be the earliest affected region (Higashi et al., 2007; Hu et al., 2008; Arai et al., 2009) followed by the entorhinal cortex and hippocampus, occipitotemporal cortex, insular and inferior temporal cortex, brainstem, and frontal neocortex and basal ganglia (Josephs et al., 2014, 2016); a scheme that has been independently validated (Tan et al., 2015). TDP-immunoreactive lesions have also been described in cognitively normal individuals (Wilson et al., 2011; Arnold et al., 2013; Uchino et al., 2015; Wennberg et al., 2019) including those with asymptomatic definite primary age related tauopathy (PART) (Josephs et al., 2017; Zhang et al., 2019), as well being associated with other well-defined clinic-pathological entities (Fig. 1) including Lewy body disease (with or without co-existing Alzheimer’s disease) (Arai et al., 2009; McAleese et al., 2017), the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (Hasegawa et al., 2007; Geser et al., 2008), Pick’s disease (Freeman et al., 2008), progressive supranuclear palsy (Yokota et al., 2010; Koga et al., 2017), corticobasal degeneration (Uryu et al., 2008; Koga et al., 2018), polyglutamine diseases such as Huntington’s disease (Schwab et al., 2008), Machado-Joseph disease (Tan et al., 2009) and spinocerebellar ataxia type 2 (Toyoshima et al., 2011), Perry syndrome (Wider et al., 2009), and chronic traumatic encephalopathy (McKee et al., 2010).
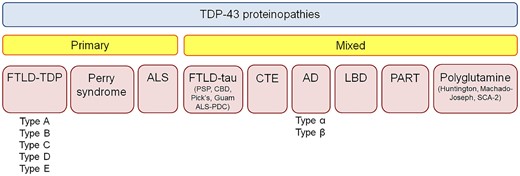
Figure 1
TDP-43 immunoreactive inclusions can be found in many different neurodegenerative diseases. Currently TDP-43 proteinopathies are divided into primary TDP-43 proteinopathies and mixed (secondary) TDP-43 proteinopathies. AD = Alzheimer’s disease; ALS = amyotrophic lateral sclerosis; CBD = corticobasal degeneration; CTE = chronic traumatic encephalopathy; FTLD = frontotemporal lobar degeneration; LBD = Lewy body disease; PART = primary age related tauopathy; PDC = Parkinson-dementia complex; PSP = progressive supranuclear palsy; SCA = spinocerebellar ataxia.
The term LATE is proposed as a catchy acronym to describe the presence of TDP-immunoreactive lesions in Alzheimer’s disease, as well as in older adults. The review manuscript describing LATE, of which many of the authors of this response are cited in, provides a thorough appraisal of an important topic and is meritorious in promoting recognition of TDP-43 and encouraging future research, as well as the development of neuroimaging and molecular biomarkers. However, we question the term’s novelty and nosology, the framework that seemingly separates LATE from FTLD-TDP and other diseases, and the proposed guidelines provided for assessing LATE and LATE-NC. As the authors identify, the term LATE is new, although there is an extensive literature on the relationship between TDP-immunoreactive inclusions and clinical and imaging features, both in isolation and in association with Alzheimer’s disease. LATE is used to rebrand already characterized features, yet identifies no new TDP-43 pathological subtype, no link between TDP-immunoreactive pathology with new cognitive symptoms (other than likelihood of dementia diagnosis) according to known brain–behaviour relationship, and no biochemical demonstration that TDP-immunoreactive lesions in older adults with and without Alzheimer’s disease are equivalent to each other or distinct from those in FTLD-TDP and other neurodegenerative diseases. Critically, using the term encephalopathy (‘E’ of LATE) presumes causation of functional impairment in the cerebrum and by incorporating the term encephalopathy, LATE implies a clinicopathological entity, when in reality LATE is only describing the pathology LATE-NC; similar to pathologies such as argyrophilic grains disease (Braak and Braak, 1987), primary age-related tauopathy (Crary et al., 2014), ageing-related tau astrogliopathy (Kovacs et al., 2016) and amygdala Lewy bodies (Uchikado et al., 2006), which are not considered clinicopathological entities. Akin to the dichotomy of language between frontotemporal dementia and FTLD, LATE is something that, by definition, cannot be diagnosed under the microscope (i.e. encephalopathy). And as these lesions can be observed in the absence of cognitive alterations, rendering a diagnosis of an encephalopathy in a cognitively normal individual is an oxymoron (normal cognition with limbic associated TDP-43 encephalopathy).
Proposing that LATE is a distinct clinicopathological entity also overlooks the possibility that TDP-immunoreactive lesions in Alzheimer’s disease could reflect impaired cellular function in end-stage neurodegeneration, and ignores the fact that the statistical association between TDP-immunoreactive pathology and a dementia diagnosis in old age is likely both not independent from Alzheimer’s disease and other pathologies, and, in all likelihood, a reflection of competing relative risks of various degrees as one ages. In addition, the term limbic-predominant is an over simplification of the distribution of pathology. In fact, it has been demonstrated that there are different subtypes of TDP immunoreactivity that have different regional associations, with only one specific subtype of neurofibrillary tangle-associated TDP proving to be truly limbic-predominant (Amador-Ortiz et al., 2007; Josephs et al., 2019; Zhang et al., 2019). The other subtype is associated with TDP-43 involving cortical and subcortical and brainstem regions identical to those affected in FTLD-TDP (Josephs et al., 2019). This is akin to Lewy body disease, which can also have a limbic-predominance, but to label it as such would ignore the brainstem and diffuse distributions of the pathology. Different subtypes of TDP-immunoreactive also have different genetic and pathological associations, including with hippocampal sclerosis; important differences that would be lost when grouping subtypes together (Murray et al., 2014; Josephs et al., 2019).
Third, this paper does not consider how LATE can be distinguished from FTLD-TDP or the fact that LATE depends on a relative selection bias towards Alzheimer’s disease-related diseases while ignoring the fact that TDP-43 has been identified in many other neurodegenerative diseases (Fig. 1) making teasing apart coincident versus associated processes a challenge. Of importance is the fact that a proportion of cases of Alzheimer’s disease and ageing have TDP-immunoreactive lesions present in frontotemporal neocortex (Arai et al., 2009; Josephs et al., 2014, 2016; Nag et al., 2018). It has been shown that such cases are strongly associated with single nucleotide polymorphisms in the TMEM106B gene (Josephs et al., 2019), which is similar to the observed associations in FTLD-TDP (Van Deerlin et al., 2010). It is, therefore, unclear whether it is appropriate to make a diagnosis of LATE in the presence of a TDP-43 stage >3 (Josephs et al., 2014, 2016), as opposed to a diagnosis of FTLD-TDP. The description of LATE also does not square with evidence that FTLD-TDP can occur in old age (Jellinger, 2006; Pao et al., 2011), and that age itself may modify the clinical phenomenology of neuropathology due to the brain’s structural and functional reorganization.
The authors provide guidelines on how to assess LATE-NC neuropathologically and suggest analysing a few specific regions of the brain with the intent to have a limited number of TDP-43 stages. While this simplifies neuropathological analysis and is touted as being cost effective, it collapses data-driven staging schemes but without the requisite scientific evidence for doing so. This proposal mirrors the NIA-AA neuropathological guidelines for the pathological diagnosis of Alzheimer’s disease, which collapse the Braak (Braak and Braak, 1991), Thal (Thal et al., 2002) and CERAD (Mirra et al., 1991) staging schemes. However, collapsing the TDP-43 staging scheme at such an early point in time when it was only published in the past few years, stifles the fields ability to understand the implications of the distribution of TDP-43 deposition and goes against the drive by many neuropathologists to include more comprehensive and quantitative assessment methods to unravel currently hidden relationships of pathologies.
The branding of clinical trials since the 1990s—especially names such as EPIC or EXCITE—is said to promote reference to such trials (Berkwits, 2000), but can compete with the understanding of the main message (Berlin, 2013; Narod et al., 2016; Witteman et al., 2018). Whether an acronym such is LATE is needed for diagnostic accuracy and communication between neuropathologists, neurologists, and other investigators is not clear. Therefore, we urge researchers to focus on defining the pathological processes and their biochemical differences underlying TDP-immunoreactive lesions in FTLD and non-FTLD disorders, particularly in diverse populations, and defer broad usage of LATE until (and only if) the science is mature.
Data availability
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Competing interests
The authors report no competing interests.
References
Amador-Ortiz
C
, Lin
WL
, Ahmed
Z
, Personett
D
, Davies
P
, Duara
R
et al. .
TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease
.
Ann Neurol
2007
;
61
:
435
–
45
.
Arai
T
, Hasegawa
M
, Akiyama
H
, Ikeda
K
, Nonaka
T
, Mori
H
et al. .
TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis
.
Biochem Biophys Res Commun
2006
;
351
:
602
–
11
.
Arai
T
, Mackenzie
IR
, Hasegawa
M
, Nonoka
T
, Niizato
K
, Tsuchiya
K
et al. .
Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies
.
Acta Neuropathol
2009
;
117
:
125
–
36
.
Arnold
SJ
, Dugger
BN
, Beach
TG
.
TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies
.
Acta Neuropathol
2013
;
126
:
51
–
7
.
Berkwits
M
.
Capture! Shock! Excite! Clinical trial acronyms and the “branding” of clinical research
.
Ann Intern Med
2000
;
133
:
755
–
62
.
Berlin
L
.
TAC: AOITROMJA? (the acronym conundrum: advancing or impeding the readability of medical journal articles?)
.
Radiology
2013
;
266
:
383
–
7
.
Bigio
EH
, Wu
JY
, Deng
HX
, Bit-Ivan
EN
, Mao
Q
, Ganti
R
et al. .
Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid
.
Acta Neuropathol
2013
;
125
:
463
–
5
.
Braak
H
, Braak
E
.
Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes
.
Neurosci Lett
1987
;
76
:
124
–
7
.
Braak
H
, Braak
E
.
Neuropathological staging of Alzheimer-related changes
.
Acta Neuropathol
1991
;
82
:
239
–
59
.
Crary
JF
, Trojanowski
JQ
, Schneider
JA
, Abisambra
JF
, Abner
EL
, Alafuzoff
I
et al. .
Primary age-related tauopathy (PART): a common pathology associated with human aging
.
Acta Neuropathol
2014
;
128
:
755
–
66
.
Freeman
SH
, Spires-Jones
T
, Hyman
BT
, Growdon
JH
, Frosch
MP
.
TAR-DNA binding protein 43 in Pick disease
.
J Neuropathol Exp Neurol
2008
;
67
:
62
–
7
.
Geser
F
, Winton
MJ
, Kwong
LK
, Xu
Y
, Xie
SX
, Igaz
LM
et al. .
Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam
.
Acta Neuropathol
2008
;
115
:
133
–
45
.
Hasegawa
M
, Arai
T
, Akiyama
H
, Nonaka
T
, Mori
H
, Hashimoto
T
et al. .
TDP-43 is deposited in the Guam Parkinsonism-dementia complex brains
.
Brain
2007
;
130
:
1386
–
94
.
Hasegawa
M
, Arai
T
, Nonaka
T
, Kametani
F
, Yoshida
M
, Hashizume
Y
et al. .
Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis
.
Ann Neurol
2008
;
64
:
60
–
70
.
Higashi
S
, Iseki
E
, Yamamoto
R
, Minegishi
M
, Hino
H
, Fujisawa
K
et al. .
Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies
.
Brain Res
2007
;
1184
:
284
–
94
.
Hu
WT
, Josephs
KA
, Knopman
DS
, Boeve
BF
, Dickson
DW
, Petersen
RC
et al. .
Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease
.
Acta Neuropathol
2008
;
116
:
215
–
20
.
Igaz
LM
, Kwong
LK
, Xu
Y
, Truax
AC
, Uryu
K
, Neumann
M
et al. .
Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis
.
Am J Pathol
2008
;
173
:
182
–
94
.
Jellinger
KA
.
Clinicopathological analysis of dementia disorders in the elderly: an update
.
J Alzheimers Dis
2006
;
9
:
61
–
70
.
Josephs
KA
, Murray
ME
, Tosakulwong
N
, Weigand
SD
, Serie
AM
, Perkerson
RB
et al. .
Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains
.
Acta Neuropathol
2019
;
137
:
227
–
38
.
Josephs
KA
, Murray
ME
, Tosakulwong
N
, Whitwell
JL
, Knopman
DS
, Machulda
MM
et al. .
Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART)
.
Acta Neuropathol
2017
;
133
:
705
–
15
.
Josephs
KA
, Murray
ME
, Whitwell
JL
, Parisi
JE
, Petrucelli
L
, Jack
CR
et al. .
Staging TDP-43 pathology in Alzheimer’s disease
.
Acta Neuropathol
2014
;
127
:
441
–
50
.
Josephs
KA
, Murray
ME
, Whitwell
JL
, Tosakulwong
N
, Weigand
SD
, Petrucelli
L
et al. .
Updated TDP-43 in Alzheimer’s disease staging scheme
.
Acta Neuropathol
2016
;
131
:
571
–
85
.
Josephs
KA
, Stroh
A
, Dugger
B
, Dickson
DW
.
Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes
.
Acta Neuropathol
2009
;
118
:
349
–
58
.
Koga
S
, Kouri
N
, Walton
RL
, Ebbert
MTW
, Josephs
KA
, Litvan
I
et al. .
Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype
.
Acta Neuropathol
2018
;
136
:
389
–
404
.
Koga
S
, Sanchez-Contreras
M
, Josephs
KA
, Uitti
RJ
, Graff-Radford
N
, van Gerpen
JA
et al. .
Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy
.
Mov Disord
2017
;
32
:
246
–
55
.
Kovacs
GG
, Ferrer
I
, Grinberg
LT
, Alafuzoff
I
, Attems
J
, Budka
H
et al. .
Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy
.
Acta Neuropathol
2016
;
131
:
87
–
102
.
Laferriere
F
, Maniecka
Z
, Perez-Berlanga
M
, Hruska-Plochan
M
, Gilhespy
L
, Hock
EM
et al. .
TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates
.
Nat Neurosci
2019
;
22
:
65
–
77
.
Lee
EB
, Lee
VM
, Trojanowski
JQ
.
Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration
.
Nat Rev Neurosci
2011
;
13
:
38
–
50
.
Lee
EB
, Porta
S
, Michael Baer
G
, Xu
Y
, Suh
E
, Kwong
LK
et al. .
Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration
.
Acta Neuropathol
2017
;
134
:
65
–
78
.
Mackenzie
IR
, Baborie
A
, Pickering-Brown
S
, Du Plessis
D
, Jaros
E
, Perry
RH
et al. .
Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype
.
Acta Neuropathol
2006
;
112
:
539
–
49
.
Mackenzie
IR
, Neumann
M
, Baborie
A
, Sampathu
DM
, Du Plessis
D
, Jaros
E
et al. .
A harmonized classification system for FTLD-TDP pathology
.
Acta Neuropathol
2011
;
122
:
111
–
3
.
McAleese
KE
, Walker
L
, Erskine
D
, Thomas
AJ
, McKeith
IG
, Attems
J
.
TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing
.
Brain Pathol
2017
;
27
:
472
–
9
.
McKee
AC
, Gavett
BE
, Stern
RA
, Nowinski
CJ
, Cantu
RC
, Kowall
NW
et al. .
TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy
.
J Neuropathol Exp Neurol
2010
;
69
:
918
–
29
.
Mirra
SS
, Heyman
A
, McKeel
D
, Sumi
SM
, Crain
BJ
, Brownlee
LM
et al. .
The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease
.
Neurology
1991
;
41
:
479
–
86
.
Murray
ME
, Cannon
A
, Graff-Radford
NR
, Liesinger
AM
, Rutherford
NJ
, Ross
OA
et al. .
Differential clinicopathologic and genetic features of late-onset amnestic dementias
.
Acta Neuropathol
2014
;
128
:
411
–
21
.
Nag
S
, Yu
L
, Boyle
PA
, Leurgans
SE
, Bennett
DA
, Schneider
JA
.
TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease
.
Acta Neuropathol Commun
2018
;
6
:
33
.
Narod
SA
, Ahmed
H
, Akbari
MR
.
Do acronyms belong in the medical literature? A countercurrents series
.
Curr Oncol
2016
;
23
:
295
–
6
.
Nelson
PT
, Dickson
DW
, Trojanowski
JQ
, Jack
CR
, Boyle
P
, Arfanakis
K
.
Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report
.
Brain
2019
;
142
:
1503
–
27
.
Nelson
PT
, Trojanowski
JQ
, Abner
EL
, Al-Janabi
OM
, Jicha
GA
, Schmitt
FA
et al. .
New old pathologies: AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS)
.
J Neuropathol Exp Neurol
2016
;
75
:
482
–
98
.
Neumann
M
, Sampathu
DM
, Kwong
LK
, Truax
AC
, Micsenyi
MC
, Chou
TT
et al. .
Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis
.
Science
2006
;
314
:
130
–
3
.
Pao
WC
, Dickson
DW
, Crook
JE
, Finch
NA
, Rademakers
R
, Graff-Radford
NR
.
Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically
.
Alzheimer Dis Assoc Disord
2011
;
25
:
364
–
8
.
Sampathu
DM
, Neumann
M
, Kwong
LK
, Chou
TT
, Micsenyi
M
, Truax
A
et al. .
Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies
.
Am J Pathol
2006
;
169
:
1343
–
52
.
Schwab
C
, Arai
T
, Hasegawa
M
, Yu
S
, McGeer
PL
.
Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease
.
J Neuropathol Exp Neurol
2008
;
67
:
1159
–
65
.
Tan
RH
, Kril
JJ
, Fatima
M
, McGeachie
A
, McCann
H
, Shepherd
C
et al. .
TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes
.
Brain
2015
;
138
:
3110
–
22
.
Tan
CF
, Yamada
M
, Toyoshima
Y
, Yokoseki
A
, Miki
Y
, Hoshi
Y
et al. .
Selective occurrence of TDP-43-immunoreactive inclusions in the lower motor neurons in Machado-Joseph disease
.
Acta Neuropathol
2009
;
118
:
553
–
60
.
Thal
DR
, Rub
U
, Orantes
M
, Braak
H
.
Phases of A beta-deposition in the human brain and its relevance for the development of AD
.
Neurology
2002
;
58
:
1791
–
800
.
Toyoshima
Y
, Tanaka
H
, Shimohata
M
, Kimura
K
, Morita
T
, Kakita
A
et al. .
Spinocerebellar ataxia type 2 (SCA2) is associated with TDP-43 pathology
.
Acta Neuropathol
2011
;
122
:
375
–
8
.
Uchikado
H
, Lin
WL
, DeLucia
MW
, Dickson
DW
.
Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy
.
J Neuropathol Exp Neurol
2006
;
65
:
685
–
97
.
Uchino
A
, Takao
M
, Hatsuta
H
, Sumikura
H
, Nakano
Y
, Nogami
A
et al. .
Incidence and extent of TDP-43 accumulation in aging human brain
.
Acta Neuropathol Commun
2015
;
3
:
35
.
Uryu
K
, Nakashima-Yasuda
H
, Forman
MS
, Kwong
LK
, Clark
CM
, Grossman
M
et al. .
Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies
.
J Neuropathol Exp Neurol
2008
;
67
:
555
–
64
.
Van Deerlin
VM
, Sleiman
PM
, Martinez-Lage
M
, Chen-Plotkin
A
, Wang
LS
, Graff-Radford
NR
et al. .
Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions
.
Nat Genet
2010
;
42
:
234
–
9
.
Wennberg
AM
, Whitwell
JL
, Tosakulwong
N
, Weigand
SD
, Murray
ME
, Machulda
MM
et al. .
The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals
.
Neurobiol Aging
2019
;
77
:
26
–
36
.
Wider
C
, Dickson
DW
, Stoessl
AJ
, Tsuboi
Y
, Chapon
F
, Gutmann
L
et al. .
Pallidonigral TDP-43 pathology in Perry syndrome
.
Parkinsonism Relat Disord
2009
;
15
:
281
–
6
.
Wilson
AC
, Dugger
BN
, Dickson
DW
, Wang
DS
.
TDP-43 in aging and Alzheimer’s disease: a review
.
Int J Clin Exp Pathol
2011
;
4
:
147
–
55
.
Witteman
HO
, Chipenda Dansokho
S
, Colquhoun
H
, Fagerlin
A
, Giguere
AMC
, Glouberman
S
et al. .
Twelve lessons learned for effective research partnerships between patients, caregivers, clinicians, academic researchers, and other stakeholders
.
J Gen Intern Med
2018
;
33
:
558
–
62
.
Yokota
O
, Davidson
Y
, Bigio
EH
, Ishizu
H
, Terada
S
, Arai
T
et al. .
Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy
.
Acta Neuropathol
2010
;
120
:
55
–
66
.
Zhang
X
, Sun
B
, Wang
X
, Lu
H
, Shao
F
, Rozemuller
AJM
et al. .
Phosphorylated TDP-43 staging of primary age-related tauopathy
.
Neurosci Bull
2019
;
35
:
183
–
92
.
Zhang
YJ
, Xu
YF
, Cook
C
, Gendron
TF
, Roettges
P
, Link
CD
et al. .
Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity
.
Proc Natl Acad Sci U S A
2009
;
106
:
7607
–
12
.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited. For commercial re-use, please contact
[email protected]







{kind=link}