




Sir,
The article recently published by Ylikallio et al. (2017) highlights mutations in the mini chromosome maintenance complex component 3 associated protein gene (MCM3AP; OMIM 603294) as a new cause of autosomal recessive Charcot-Marie-Tooth (CMT) neuropathy associated with intellectual disability (Ylikallio et al., 2017). The authors identified four compound heterozygous mutations (p.Pro148Leufs*48/p.Val1272Met; p.Tyr889*/p.Ala867Asp; p.Gln619=/p.Asn1317fs*18; p.Ser1478*/p.Glu1577Lys) and a homozygous missense mutation (p.Met762Thr) in five families of different ethnicity. Three of the mutant alleles were frameshift nonsense mutations in which one was de novo (p.Tyr889*). One of the mutant alleles was a synonymous mutation (p.Gln619=) located in the splice donor position of exon 5 and predicted to create an exonic splicing silencer site. Since the article of Ylikallio et al., two additional homozygous mutations have been reported in Kurdish (p.Arg878His) and Iranian (p.Ser951Pro) consanguineous families and a compound heterozygous mutation (Gln1804*/Ser145_Leu1459dup) in a non-consanguineous Iranian family (Karakaya et al., 2017). Prior to the report of this gene for recessive CMT, studies of families with intellectual disability described two siblings with a homozygous variant in the MCM3AP gene (p.Glu915Lys). Interestingly, this gene was selected as a candidate gene in which the siblings presented with borderline to mild intellectual disability, progressive polyneuropathy and ptosis (Schuurs-Hoeijmakers et al., 2013).
We would like to report the use of whole exome sequencing to identify a novel missense mutation (NM_003906.3 c.2609T>C p.Leu870Ser; chr21:g.47690334) in exon 9 of the MCM3AP gene in three siblings (two sisters and one brother) of Lebanese ethnicity. The onset of slowly progressive distal muscle weakness involving wrists, hand intrinsic muscles, feet and ankles occurred between ages 6 and 10 years. Early motor milestones had been normal but by early teens all had developed foot drop, wrist and finger drop and were significantly disabled. The parents were clinically normal and distantly related. One additional brother had no neurological deficits and was unavailable for testing. The three affected siblings were homozygous for the mutation and both parents were heterozygous carriers for the p.Leu870Ser mutant allele (Fig. 1A).
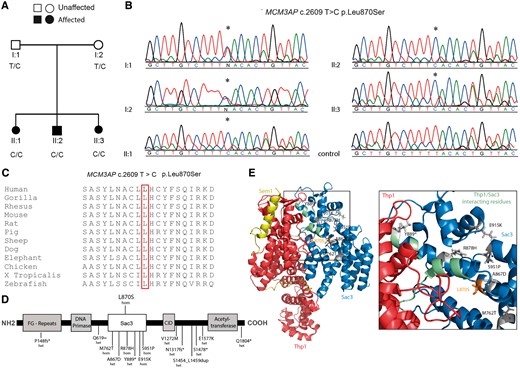
The p.Leu870Ser MCM3AP mutation in a Lebanese family localizes to the Sac3 domain of GANP. (A) The pedigree shows segregation of the mutation that has been confirmed by (B) Sanger sequencing analysis traces. Circles denote females and squares denote males. The asterisk denotes the nucleotide position at which a homozygous T to C transition is observed in all affected siblings and both parents are heterozygous for the T to C transition. A sequence trace of a neurologically normal unrelated individual shows a homozygous T allele. (C) Alignment analysis for the p.Leu870Ser mutation for MCM3AP orthologues in different species. Amino acid position 870 is boxed in red. (D) GANP protein showing the different domains and location of mutations for recessive CMT. Previously reported mutations (below) and the novel mutation in this study (above) are shown. (E) Structure of the TREX2 complex including the GANP protein, showing CMT causing mutations within the Sac3 domain. Diagram was drawn using the crystal structure of the S. cerevisiae homologue (PDB: 3t5v) (Ellisdon et al., 2012). Sac3 residues are shown in blue, in complex with Thp1 (human homologue PCID2) in red and Sem1 (human homologue DSS1) in yellow. CMT mutations are shown as grey sticks and the newly identified p.Leu870Ser mutation in orange. Box area is enlarged to show CMT mutations enriched around the Sac3/Thp1 interaction interface (Sac3 interacting residues highlighted in green).
The clinical features of the affected family members are shown in Table 1. When first reviewed at the Concord Hospital Adult Neuromuscular Clinic aged in their early 20 s, the affected siblings all had marked wasting of hand intrinsic and forearm muscles and wasting of the foot intrinsic and leg anterior and posterior compartment muscles. Proximal muscle bulk was normal. All had grade 1–2 power in hand intrinsic muscles and finger extensors with grade 4 strength in long finger flexors and wrist flexors. Proximal upper limb power remained normal. In the legs power was graded as ankle dorsiflexion 2, peronei 2, tibialis posterior 4, plantar flexion 4+ with normal power proximally. Upper limb and knee tendon reflexes were normal and ankle jerks could just be elicited. All three had reduced sensation to pain and temperature to the mid forearm and mid-thigh. All had intact vibration and proprioception. The affected siblings were unsteady but independently mobile with bilateral foot drop and frequent falls. All married in their early 20 s and have managed to maintain independent gait and continued to manage their activities of daily living with assistance from their spouses and parents. Each has had more than one child all of whom remain normal. All of the affected siblings have become increasing obese and two have required treatment for diabetes mellitus type 2 with oral agents. All three have developed obstructive sleep apnoea but no dysfunction of bulbar musculature.
Summary of clinical features of the patients
| Patient | II:1 | II:2 | II:3 |
|---|---|---|---|
| Gender | F | M | F |
| First evaluation age | 23 | 25 | 21 |
| UL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| LL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| Bilateral foot drop | +++ | +++ | +++ |
| Sensory loss | Distally to pain | Distally to pain | Distally to pain |
| Nerve conduction studies age | 24 years | 26 years | |
| Median motor, mV/m/s | Absent | Absent | |
| Ulnar motor, mV/m/s | 0.6/38.4 | Not recorded | 0.3 |
| Tibial motor, mV/m/s | 2.4 | Not recorded | 4.2/43 |
| Common peroneal motor, mV/m/s | Absent | Absent | |
| Median sensory, µV/m/s | 7.3/49.8 | Not recorded | 6.9/48 |
| Ulnar, µV/m/s | 3.7/52.6 | Not recorded | 4/52.4 |
| Sural, µV/m/s | Not recorded | Not recorded | 5.1/48 |
| Posterior tibial evoked potentials | Normal | Not recorded | Normal |
| Brain MRI | Middle fossa arachnoid cysts; otherwise normal | Normal | |
| Spinal MRI | Normal | ||
| Additional medical problems | Obesity, DM type 2, psychosis | Obesity | Obesity, epigastric hernia repair, DM type 2 |
| Patient | II:1 | II:2 | II:3 |
|---|---|---|---|
| Gender | F | M | F |
| First evaluation age | 23 | 25 | 21 |
| UL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| LL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| Bilateral foot drop | +++ | +++ | +++ |
| Sensory loss | Distally to pain | Distally to pain | Distally to pain |
| Nerve conduction studies age | 24 years | 26 years | |
| Median motor, mV/m/s | Absent | Absent | |
| Ulnar motor, mV/m/s | 0.6/38.4 | Not recorded | 0.3 |
| Tibial motor, mV/m/s | 2.4 | Not recorded | 4.2/43 |
| Common peroneal motor, mV/m/s | Absent | Absent | |
| Median sensory, µV/m/s | 7.3/49.8 | Not recorded | 6.9/48 |
| Ulnar, µV/m/s | 3.7/52.6 | Not recorded | 4/52.4 |
| Sural, µV/m/s | Not recorded | Not recorded | 5.1/48 |
| Posterior tibial evoked potentials | Normal | Not recorded | Normal |
| Brain MRI | Middle fossa arachnoid cysts; otherwise normal | Normal | |
| Spinal MRI | Normal | ||
| Additional medical problems | Obesity, DM type 2, psychosis | Obesity | Obesity, epigastric hernia repair, DM type 2 |
DM = diabetes mellitus; F = female; LL = lower limb; M = male; UL = upper limb. − = normal power; +++ = less than antigravity power .
Summary of clinical features of the patients
| Patient | II:1 | II:2 | II:3 |
|---|---|---|---|
| Gender | F | M | F |
| First evaluation age | 23 | 25 | 21 |
| UL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| LL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| Bilateral foot drop | +++ | +++ | +++ |
| Sensory loss | Distally to pain | Distally to pain | Distally to pain |
| Nerve conduction studies age | 24 years | 26 years | |
| Median motor, mV/m/s | Absent | Absent | |
| Ulnar motor, mV/m/s | 0.6/38.4 | Not recorded | 0.3 |
| Tibial motor, mV/m/s | 2.4 | Not recorded | 4.2/43 |
| Common peroneal motor, mV/m/s | Absent | Absent | |
| Median sensory, µV/m/s | 7.3/49.8 | Not recorded | 6.9/48 |
| Ulnar, µV/m/s | 3.7/52.6 | Not recorded | 4/52.4 |
| Sural, µV/m/s | Not recorded | Not recorded | 5.1/48 |
| Posterior tibial evoked potentials | Normal | Not recorded | Normal |
| Brain MRI | Middle fossa arachnoid cysts; otherwise normal | Normal | |
| Spinal MRI | Normal | ||
| Additional medical problems | Obesity, DM type 2, psychosis | Obesity | Obesity, epigastric hernia repair, DM type 2 |
| Patient | II:1 | II:2 | II:3 |
|---|---|---|---|
| Gender | F | M | F |
| First evaluation age | 23 | 25 | 21 |
| UL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| LL weakness proximal/distal | −/+++ | −/+++ | −/+++ |
| Bilateral foot drop | +++ | +++ | +++ |
| Sensory loss | Distally to pain | Distally to pain | Distally to pain |
| Nerve conduction studies age | 24 years | 26 years | |
| Median motor, mV/m/s | Absent | Absent | |
| Ulnar motor, mV/m/s | 0.6/38.4 | Not recorded | 0.3 |
| Tibial motor, mV/m/s | 2.4 | Not recorded | 4.2/43 |
| Common peroneal motor, mV/m/s | Absent | Absent | |
| Median sensory, µV/m/s | 7.3/49.8 | Not recorded | 6.9/48 |
| Ulnar, µV/m/s | 3.7/52.6 | Not recorded | 4/52.4 |
| Sural, µV/m/s | Not recorded | Not recorded | 5.1/48 |
| Posterior tibial evoked potentials | Normal | Not recorded | Normal |
| Brain MRI | Middle fossa arachnoid cysts; otherwise normal | Normal | |
| Spinal MRI | Normal | ||
| Additional medical problems | Obesity, DM type 2, psychosis | Obesity | Obesity, epigastric hernia repair, DM type 2 |
DM = diabetes mellitus; F = female; LL = lower limb; M = male; UL = upper limb. − = normal power; +++ = less than antigravity power .
Two of the affected siblings have had one or more nerve conduction studies. At age 26 years the youngest daughter (Patient II:3) had nerve conduction studies demonstrating normal median, ulnar and sural sensory nerve action potentials. The motor compound action potentials were absent or markedly reduced in the upper limbs and reduced but less affected in the legs. Motor conduction velocities were reduced but sensory velocities normal. For the oldest sibling (Patient II:1) at age 24 years, sensory nerve action potentials could be elicited but the amplitudes were reduced. Motor compound action potentials were reduced or absent, terminal motor latencies normal and motor conduction velocities reduced. By age 28 there was a significant deterioration in both motor and sensory parameters with sural sensory potentials no longer recordable. Needle EMG demonstrated active and chronic neurogenic changes. Posterior tibial nerve somatosensory evoked potentials recorded in the early 20 s for Patients II:1 and II:3 demonstrated normal lumbar and cerebral potential waveforms and latencies. None of the siblings experienced significant pain, sensory symptoms or cramping. All were able to walk independently and have used ankle foot orthoses intermittently. Because of severe hand weakness they were not able to use their hands to hold walking aids. The eldest sibling (Patient II:1) developed psychosis with catatonia requiring treatment with electroconvulsive therapy subsequently completely controlled.
The p.Leu870Ser mutation we report was absent from public databases including dbSNP 150, gnomAD, 1000 genomes and the Greater Middle East Variome Project (GME Variome). The variant was also excluded from 200 neurologically normal chromosomes (n = 100 in-house controls). The amino acid is highly conserved and a suite of prediction tools suggest the mutation is disease causing (Fig. 1C and Supplementary Table 1). As reported for the other MCM3AP homozygous mutations, the CADD score for p.Leu870Ser (29.0) was >20 indicating the substitution is in the top 1% most deleterious changes to occur in the genome (Kircher et al., 2014).
As noted in previous studies, the genotype–phenotype correlation of patients with MCM3AP mutations is unclear. With the addition of the p.Leu870Ser mutation, there are now five homozygous mutations reported. Interestingly, the homozygous mutations are all located in the suppressor of actin 3 (Sac3) domain (amino acid 366–990) of GANP. Patients presenting with homozygous mutations all present with debilitating neuropathy with two mutations (p.Met762Thr and p.Glu915Lys) associated with intellectual disability.
The MCM3AP gene encodes the germinal centre associated nuclear protein (GANP) and is ubiquitously expressed (Abe et al., 2000). The nuclear phase of the gene expression pathway is the export of mature mRNAs to the cytoplasm through nuclear pore complexes. GANP is a subunit of TREX-2, which is an important mRNA export complex (Jani et al., 2012). All the homozygous mutations reported to date cluster in the Sac3 domain (Fig. 1D). Modelling the mutations in the crystal structure of the Saccharomyces cerevisiae homologue (Ellisdon et al., 2012) reveals that they lie in close proximity to the Sac3-ThpI (human PCID2) binding interface (Fig. 1E). Maintenance of this interface is needed for mRNA export through the nuclear pore complexes. It is therefore possible that our mutation and others reported within this domain could affect mRNA export by disturbing the Sac3-PCID2 interface; however, further functional studies will be required to confirm this hypothesis. Mutations in the MCM3AP gene highlight the importance of understanding mRNA export in the pathogenic process of axonal degeneration that underlies CMT.
Web resources
The URLs used in this study: Online Mendelian Inheritance in Man; https://www.omim.org/ UCSC Genome Browser; https://genome.ucsc.edu/ Combined Annotation Dependent Depletion (CADD; http://cadd.gs.washington.edu/home Genome Aggregation Database gnomAD; http://gnomad.broadinstitute.org/ 1000 Genomes Browser; http://www.internationalgenome.org/1000-genomes-browsers/ The Greater Middle East Variome Project GME; http://igm.ucsd.edu/gme/ Mutation Taster; http://www.mutationtaster.org/ Polyphen; http://genetics.bwh.harvard.edu/pph2/ SIFT and Provean; http://sift.jcvi.org/ Clustal Omega; https://www.ebi.ac.uk/Tools/msa/clustalo/
Acknowledgements
We would like to thank the family members for participating in this study.
Funding
This work was supported by the Australian National Health and Medical Research Council project grant (APP1046690) awarded to M.L.K and G.A.N.


{kind=link}