




Sir,
In a recent investigation Deshaies and colleagues (2018) identified HNRNP A1-7B, an isoform of the RNA binding protein (RBP) HNRNP A1, as present in post-mortem amyotrophic lateral sclerosis (ALS) spinal motor neuron inclusions. HNRNP A1 is an important player in ALS, as mutations in its low complexity domain (LCD) can be causative for the disease (Kim et al., 2013). Intriguingly, the isoform described by Deshaies et al. includes exon 7B, which produces an extension of the LCD by 52 amino acids, pointing to a link between this alternative splicing event and neuronal protein aggregation. The authors further suggest that loss of RBP TDP-43 can influence this splicing event and result in an increase of the aggregation-prone 7B isoform. TDP-43 is abnormally mislocalized in >95% of ALS cases and, when mutated, can cause ALS (Harrison and Shorter, 2017).
TDP-43 binds RNA, preferentially at UG repeats, and is involved in alternative splicing (Buratti and Baralle, 2001). Importantly, its activity is extremely dosage-sensitive, making physiological expression an essential condition for studying the effect of TDP-43 mutations on splicing. Recently, we and others have published an allelic series of novel physiological TDP-43 mouse mutant models and shown that C-terminal mutations induce RNA splicing gain-of-function (Fratta et al., 2018; White et al., 2018). To investigate the link between TDP-43, its mutations and Hnrnpa1 exon 7B splicing, we used recently published high quality iCLIP (Tollervey et al., 2011; Rogelj et al., 2012) and RNA-seq datasets (Kapeli et al., 2016; Fratta et al., 2018; White et al., 2018) from human and mouse.
Deshaies et al. show, by immunoprecipitation of TDP-43 followed by RT-PCR, that TDP-43 directly interacts with HNRNPA1 transcripts. However, their approach does not allow identification of the exact site of interaction (Deshaies et al., 2018). To determine the exact binding sites, we pooled available mouse and human iCLIP data for TDP-43, which identifies the binding sites of TDP-43 on transcripts with single nucleotide resolution. We found that TDP-43 does not bind to exon 7B or its flanking introns, where UG rich motifs are present, but instead binds to HNRNPA1 exon 8 (in human) and has very strong conserved binding sites within the 3′UTR of HNRNPA1 in both human and mouse (Fig. 1A).
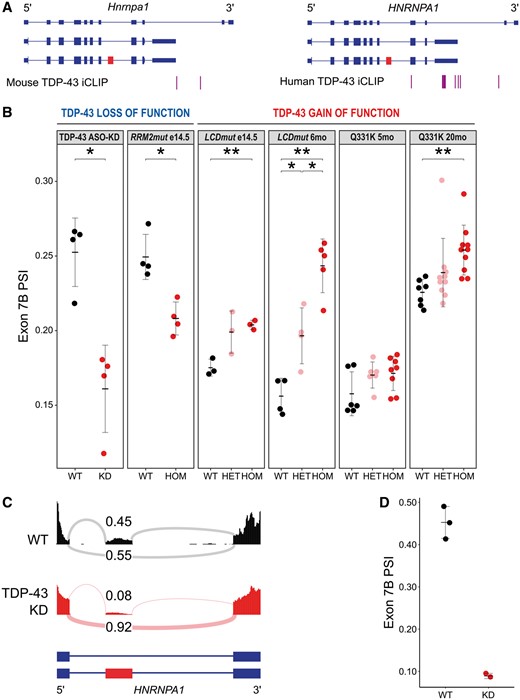
HNRNP A1 isoforms in TDP-43 loss- and gain-of-function models. (A) Annotated Hnrnpa1 transcripts alongside iCLIP clusters indicating regions of TDP-43 binding in mouse (left) and human (right). Exon 7B highlighted in red. (B) Percentage spliced in (PSI) values for Hnrnpa1 exon 7B in six published RNA-seq datasets: TDP-43 ASO-KD, RRM2mut embryonic Day 14.5 frontal cortex, LCDmut embryonic Day 14.5 head, LCDmut 6-month spinal cord, Q331K 5-month spinal cord and Q331K 20-month spinal cord. *P < 0.05, **P < 0.01. (C) Representative RNA-seq read traces with sashimi plots for HNRNPA1 in wild-type (WT) and TDP-43 KD human iMNs. Proportional levels of exon 7B inclusion (top) and skipping (bottom) labelled in each trace. Exon 7B highlighted in red on annotated HNRNPA1 transcripts. (D) PSI values for HNRNPA1 exon 7B in TDP-43 KD human iMN RNA-seq. RNA-Seq dataset analysis and PSI calculation were conducted as described in Fratta et al. (2018).
To investigate the effect of TDP-43 loss-of-function in mouse CNS, we used two RNA-seq datasets where: (i) TDP-43 knockdown through antisense oligonucleotides reduces levels of TDP-43 in the adult mouse brain (TDP-43 ASO-KD) (Polymenidou et al., 2011); and (ii) point mutation in TDP-43 RNA recognition motif 2 (RRM2) reduces its RNA binding capacity and therefore uncovers direct consequences of diminished TDP-43 binding to RNA (TDP-43 RRM2mut) (Fratta et al., 2018).
Strikingly, both TDP-43 ASO-KD and TDP-43 RRM2mut displayed greater skipping of Hnrnpa1 exon 7B (Fig. 1B), rather than the greater inclusion levels described by Deshaies et al. We then analysed the impact of C-terminal mutations in TDP-43 upon Hnrnpa1 and its splicing. One mutation, TDP-43 Q331K, is causative for ALS in patients and has been shown to induce cognitive phenotypes in mice; the second mutation, TDP-43 LCDmut, has not been described in patients but lies within the hotspot for human TDP-43 ALS mutations and induces an ALS-like phenotype in mice (Fratta et al., 2018; White et al., 2018). Recent work on both lines has demonstrated a gain of splicing function effect of C-terminal TDP-43 mutants. Intriguingly, both mutations show a dose-dependent increase in Hnrnpa1 exon 7B inclusion (Fig. 1B), the opposite of what occurs in TDP-43 RRM2mut and TDP-43 ASO-KD. Both TDP-43 LCDmut and Q331K models, which show increased Hnrnpa1 exon 7B inclusion, develop a clear neurodegenerative disease phenotype (Fratta et al., 2018; White et al., 2018). This link agrees with Deshaies et al.’s finding that HNRNP A1-7B accumulates in ALS patient spinal cord motor neurons. Interestingly, in both TDP-43 LCDmut and the Q331K models, the increased exon 7B inclusion is detectable at all timepoints, but is more pronounced at late disease stages.
Finally, we examined whether the effects of changes in TDP-43 splicing activity on HNRNPA1 exon 7B inclusion were conserved in humans. We analysed RNA-seq data from shRNA-mediated TDP-43 knockdown in motor neurons generated from wild-type human iPSCs (iMNs) (Kapeli et al., 2016). In accordance with results from our mouse model analysis, loss of TDP-43 in human settings also resulted in substantial reduction in exon 7B inclusion (Fig. 1C and D).
In summary, our data here support the direct involvement of TDP-43 in HNRNPA1 splicing. We show that TDP-43 gain-of-function, but not loss-of-function, leads to the increased inclusion of exon 7B. Although our findings may differ from Deshaies et al. on how TDP-43 loss-of-function impacts on HNRNPA1 splicing, both our data and Deshaies et al. support a link between TDP-43, the HNRNP A1-7B isoform and ALS.
Data availability
The data that support the findings of this study are openly available in the following locations: F210I/M323K, NCBI Sequence Read Archive reference SRP133158; mouse TDP-43 KD, NCBI Sequence Read Archive reference SRP005860; Q331K, Gene Expression Omnibus reference GSE99354; human TDP-43 KD, Gene Expression Omnibus reference GSE77707.
Funding
This research was supported by the UK Medical Research Council, the UK Motor Neuron Disease Association, the Rosetrees Foundation, the Thierry Latran Foundation and the Miguel Servet Programme of the ISCii, Spain.
Competing interests
The authors report no competing interests.


{kind=link}