




Abstract
Hereditary spastic paraplegias (HSPs) are rare neurological disorders caused by progressive distal degeneration of the corticospinal tracts. Among the 79 loci and 65 spastic paraplegia genes (SPGs) involved in HSPs, mutations in SPAST, which encodes spastin, responsible for SPG4, are the most frequent cause of both familial and sporadic HSP. SPG4 is characterized by a clinically pure phenotype associated with restricted involvement of the corticospinal tracts and posterior columns of the spinal cord. It is rarely associated with additional neurological signs. However, both age of onset and severity of the disorder are extremely variable. Such variability is both intra- and inter-familial and may suggest incomplete penetrance, with some patients carrying mutations remaining asymptomatic for their entire life. We analysed a cohort of 842 patients with SPG4-HSP to assess genotype–phenotype correlations. Most patients were French (89%) and had a family history of SPG4-HSP (75%). Age at onset was characterized by a bimodal distribution, with high inter-familial and intra-familial variability, especially concerning first-degree relatives. Penetrance of the disorder was 0.9, complete after 70 years of age. Penetrance was lower in females (0.88 versus 0.94 in males, P = 0.01), despite a more diffuse phenotype with more frequent upper limb involvement. Seventy-seven per cent of pathogenic mutations (missense, frameshift, splice site, nonsense, and deletions) were located in the AAA cassette of spastin, impairing its microtubule-severing activity. A comparison of the missense and truncating mutations revealed a significantly lower age at onset for patients carrying missense mutations than those carrying truncating mutations, explaining the bimodal distribution of the age at onset. The age at onset for patients carrying missense mutations was often before 10 years, sometimes associated with intellectual deficiency. Neuropathological examination of a single case showed degeneration of the spinocerebellar and spinocortical tracts, as well as the posterior columns. However, there were numerous small-diameter processes among unusually large myelinated fibres in the corticospinal tract, suggesting marked regeneration. In conclusion, this large cohort of 842 individuals allowed us to identify a significantly younger age at onset in missense mutation carriers and lower penetrance in females, despite a more severe disorder. Neuropathology in one case showed numerous small fibres suggesting regeneration.
Introduction
Hereditary spastic paraplegias (HSPs) are inherited disorders caused by the progressive degeneration of the corticospinal tracts, leading to gait disorder and spasticity of the lower limbs. HSPs are characterized by the high heterogeneity of both their genetic background and clinical manifestation. The number of known HSP genes continues to increase, with 79 loci and 65 genes (spastic paraplegia genes, SPGs) (Parodi et al., 2018) identified to date. Pathogenic mutations can manifest sporadically or be transmitted through all known inheritance patterns (Klebe et al., 2015), mostly by autosomal-dominant and recessive transmission, accounting for an overall prevalence of 1–5:100 000 (Ruano et al., 2014). HSPs have been historically classified into ‘pure’ and ‘complex’ forms based on symptoms: pure HSPs are defined by an upper motor neuron phenotype (spastic gait, lower-limb hypertonicity, hyperreflexia, extensor plantar responses, sphincter disturbances, and proximal weakness), whereas the presence of additional neurological or extraneurological signs define the ‘complex’ or ‘complicated’ form of the disorder (Fink, 2014). Degeneration may also affect the lower motor neurons, associated with an ALS phenotype (Parodi et al., 2017). The age at onset of HSPs is variable, ranging from early childhood to 70 years of age (Salinas et al., 2008). Carriers may thus be asymptomatic for decades and the probability of being affected (penetrance) is age-dependent (Harding, 1981; Durr et al., 1993).
SPG4 is due to heterozygous mutations of the SPAST gene and is the most frequent cause of both familial and sporadic HSP (Lo Giudice et al., 2014). Located on chromosome 2p22.3, SPAST encodes spastin (Hazan et al., 1999), a protein belonging to the AAA family. Spastin hydrolyses ATP to sever microtubules and controls various aspects of microtubule dynamics, such as their number, length, and motility (Errico et al., 2002). Spastin is composed of four domains necessary for its enzymatic function and interaction with intracellular partners. It is involved in endoplasmic reticulum morphogenesis (Park et al., 2010) and lipid metabolism (Papadopoulos et al., 2015) through its N-terminal domain (starting from residue 1 to 87), together with other SPG-encoded proteins such as ATL1/SPG3A and REEP1/SPG31. The microtubule interacting and trafficking domain (MIT), located between amino acids 116 and 194, allows interactions with two proteins belonging to the endosomal-sorting complex required for transport III (ESCRT-III) machinery, CHIMP1 and IST1, explaining the role of spastin in both cytokinesis and endosomal-tubule recycling (Reid et al., 2005; Connell et al., 2009; Allison et al., 2013). The two remaining domains, the microtubule-binding domain (MTBD), comprising amino acids 270 to 328, and the AAA ATPase cassette, from amino acid 342 to 616, are crucial for spastin-severing activity. Microtubules are severed by the energy derived from ATP hydrolysis following the assembly of six spastin subunits into a ring-shaped hexamer that binds to microtubules and introduction of the C terminus of tubulin into the central pore (White et al., 2007; Roll-Mecak and Vale, 2008).
Four spastin isoforms are produced by an alternative initiation codon and differential exon 4 splicing (Havlicek et al., 2014). Spastin isofom M1 (68 kDa) and M87 (60 kDa) are produced by an alternative translation start site (Claudiani et al., 2005; Mancuso and Rugarli, 2008) and share all protein domains, except for the N-terminal domain, which is present only in the M1 isoform. Moreover, the human M87 isoform is expressed both in the spinal cord and cerebral cortex, whereas spastin-M1 is detectable only in the spinal cord (Solowska et al., 2010).
SPG4-HSP has been defined as a ‘pure’ HSP, based on the neurological symptoms presented by affected patients, with the lesions being restricted to the corticospinal tract and the posterior column (Hazan et al., 1999; Fonknechten et al., 2000). The most frequent additional observations have concerned cognitive decline and white matter abnormalities by cerebral MRI (Tallaksen et al., 2003; Nielsen et al., 2004; Ribaï et al., 2008; Murphy et al., 2009). SPG4-HSP is characterized by an age of onset ranging from childhood to the eighth decade of life. The presence of genetic modifiers underlies such high intra- and inter-familial variability, mostly affecting age of onset and severity. To date, the SPAST c.131C>T/p.(Ser44Leu) variant is the most well established SPG4-HSP genetic modifier, leading to a much lower age at onset and the expression of a more severe phenotype in carriers when associated in trans with a SPAST mutation (Svenson et al., 2004; McDermott et al., 2006).
We investigated the factors that account for phenotypic variability in detail by analysing a cohort of 842 SPG4-mutated patients recruited through the diagnostic unit of the Pitié-Salpêtrière University Hospital or the SPATAX network (www.spatax.wordpress.com). Data concerning the specific mutations, longitudinal neurological signs, the age at onset, and disorder severity were used to assess phenotype-genotype correlations and identify genetic modifiers.
Materials and methods
Patients
This study included 842 HSP-affected patients, 529 from the SPATAX network and 313 that were referred to the clinical diagnostic laboratory of the Genetics Department (G.B. and Ch. De.) of the Pitié-Salpêtrière University Hospital. Clinical information based on neurological examinations and genetic counselling were available for 636 patients.
Most of the patients had a family history of the disorder (75.6%, 481/636), whereas 36 (5.7%) were sporadic cases. Information concerning transmission of the disorder was unavailable for the remaining individuals (18.7%, 119/636).
In addition to pyramidal signs in the lower limbs, we listed the frequency of increased reflexes of the upper limbs, Hoffmann sign, and weakness and wasting of the lower limbs, as well as urinary disturbances, pes cavus, a decreased sense of vibration at the ankles, and cognitive difficulties, including intellectual deficiency and cognitive decline with age. Disorder severity was assessed using the SPATAX disability scale (0: no functional handicap; 1: no functional handicap but signs at examination; 2: mild, able to run, walking unlimited; 3: moderate, unable to run, limited walking without aid; 4: severe, walking with one cane; 5: walking with two canes; 6: unable to walk, requiring a wheelchair; 7: confined to bed). In addition, the disability progression index (ratio of disability stage and disorder duration) at the last examination was used to evaluate progression of the disorder: the difference in severity between the last and first examination and the corresponding elapsed time were taken into account to assess progression. Rapidly and slowly progressing patients were above or below the median of the disability progression index, respectively, of 0.09 in the analysed follow-up cohort.
Penetrance was calculated as the number of affected patients divided by the total number of carriers.
All patients included in the present study gave their consent for DNA testing in the research setting and consent forms (RBM 01-29, RBM 03-48) were signed by each participant.
Mutation analysis
Genomic DNA was extracted from peripheral blood and both targeted sequencing and Sanger sequencing were performed to detect mutations that potentially affect SPAST exons and portions of the neighbouring intronic regions. Multiplex ligation-dependent probe amplification (MLPA) was used to identify the extent of large deletions and intragenic SPAST rearrangements (SALSA MLPA probemix P165, MRC-Holland). MLPA results were analysed using either GeneMapper® (version 4, ThermoFisher) or Coffalyzer.net (MRC-Holland). Mutations were interpreted using Alamut 8.0 (Interactive Biosoftware, Rouen, France), which includes Align GVGD (http://agvgd.hci.utah.edu/), PolyPhen-2 [Human Diversity (HumDiv), and Human Variation (HumVar) prediction models, http://genetics.bwh.harvard.edu/pph2/], MutationTaster (http://www.mutationtaster.org/) and SIFT prediction test (http://sift.jcvi.org/). The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and the genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) were consulted for single nucleotide polymorphisms (SNPs).
Frameshift and nonsense mutations occurring in exon sequences (NM_14946.3) were considered to be deleterious. Missense mutations affecting highly conserved amino acids of the AAA domain and predicted to be deleterious by at least three in silico tools were considered to be deleterious. Missense mutations affecting a highly conserved amino acid outside the AAA domain and predicted to be deleterious by at least three in silico tools were considered to be deleterious only when supported by Sanger sequencing segregation analysis. Finally, in silico predictions of splice mutations were performed using five models: SpliceSiteFinder-like (Interactive Biosoftware), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), GeneSplicer (http://www.cbcb.umd.edu/software/GeneSplicer), and Human Splicing Finder (http://www.umd.be/HSF3/HSF.shtml).
Missense, splice-site, nonsense, and frameshift mutations were annotated using wANNOVAR (http://wannovar.wglab.org/) (Wang et al., 2010; Yang and Wang, 2015), allowing the assessment of gnomAD frequencies and mutation CADD scores (Kircher et al., 2014).
Statistical analysis
Chi-square tests of clinical data were performed using IBM SPSS Statistics (IBM Corp. Released 2013. IBM SPSS Statistics for Macintosh, Version 22.0. Armonk, NY: IBM Corp.). The means, standard deviations, Mann-Whitney tests (two-tailed), chi-square tests, and log-rank tests were computed using GraphPad Prism version 6.00 for Macintosh (GraphPad Software, La Jolla California USA, www.graphpad.com). R package ggplot2 was used for graphic representations (Wickham, 2009). The resulting P-values were considered to be statistically significant when <0.05.
The FCOR package, part of S.A.G.E software packages (S.A.G.E. [2016] Statistical Analysis for Genetic Epidemiology, Release 6.4: http://darwin.cwru.edu), was used to calculate intra-familial correlations.
Neuropathological examination
A post-mortem neuropathological examination was performed on Patient FSP-625-011, who died from brainstem compression due to cerebral haemorrhage related to vesical cancer at age 59. The brain and spinal cord were available for examination. Horizontal sections of the spinal cord were prepared and staining included haematin-eosin, Luxol fast blue for myelin, CD68 for microglia, and double labelling of myelin basic protein (MBP) and neurofilaments. The lateral column was cut sagittally, along the long axis of the spinal cord at the thoracic level, to examine the aspect of the fibres of the pyramidal tract.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data, processed in accordance with consent forms RBM 01-29 and RBM 03-48, are not publicly available due to the European General Data Protection Regulation (GDPR).
Results
Age of onset and intra-familial correlation
Age of onset could be determined for 547 patients and ranged from 0 to 77 years, with a mean age of 29 years (29.3 ± 18.6). Fifty-one mutation carriers did not report any age at onset because they felt asymptomatic (mean age at examination 37.3 ± 15.6 years, ranging from 6 to 70), but 28/51 had an extensor plantar reflex in the lower limbs. Only 23 mutations carriers, with a mean age at examination of 39.5 ± 16 years, ranging from 17 to 70 years, were asymptomatic and had no apparent pyramidal signs. Overall penetrance of the disorder was 0.9 and was complete at 70 years of age.
Index cases were distinguished from other family members to avoid possible bias and assess the distribution of age at onset. The mean age at onset was similar for the index cases (28.7 ± 17, range 0 to 69, n = 215) and family members (27.3 ± 19, range 0 to 77, n = 213; Mann-Whitney test, P = 0.38). The onset of SPG4-HSP followed a bimodal distribution among both probands and non-probands, characterized by a first peak extending from birth to the first decade and a second peak from the third to fifth decades (Fig. 1). The mean age at the last examination was 47 years (47 ± 17.8, n = 633, range 1.5 to 95). Both the age at onset and at the last examination were available for 546 patients; the mean disorder duration (age at last examination minus the age of onset) was 18 years (18.5 ± 14.7, range 6 months to 75 years).
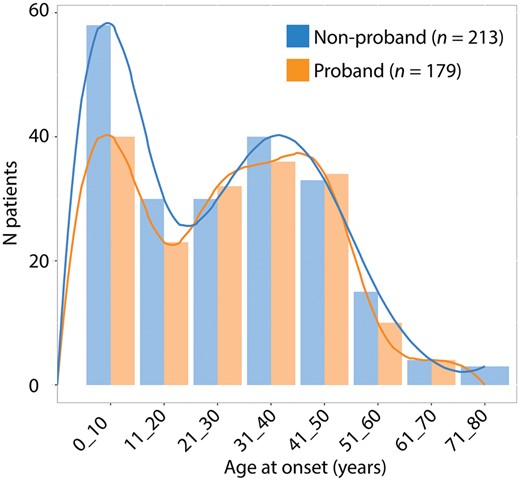
Distribution of the age at onset of the analysed cohort. Age at onset was compared among non-probands and probands. The resulting bimodal trend shows that the onset of SPG4-HSP occurs between birth and the first decade and between the third and fifth decades.
Information retrieved from 84 pedigrees with at least two affected individuals with the known age at onset were used to estimate the intra-familial correlation of age at onset. The correlation was highly significant for the 113 sibling pairs (r = 0.37, P = 0.0008) and 109 parent-offspring pairs (r = 0.34, P = 0.001) analysed. The correlation was still significant for uncle-nephew pairs (r = 0.27, n = 104, P = 0.042), but lost significance between grandparents and grandchildren (r = 0.40, n = 22, P = 0.105). The correlations were affected by gender: the correlation was highly significant between brothers (r = 0.58, n = 34, P = 0.0002), did not reach significance between sisters (r = 0.39, n = 27, P = 0.059), and was still lower for brother-sister pairs (r = 0.23, n = 52, P = 0.187).
The difference in the age of onset between members of the same family was 27 years on average (26.9 years ± 18.3, n = 28) for the 84 families of our cohort, with a maximum of 69 years separating onset of the disorder for two related patients.
Clinical signs and severity
The mean level of disability of the overall cohort was 2.95 ± 1.6 (n = 481), with only one patient reaching the highest severity level of 7. The overall clinical presentation was spasticity in the lower limbs, more pronounced at gait than at rest, which was rarely severe. Half of the cohort showed proximal weakness and increased reflexes in the upper limbs (Table 1). Oculomotor abnormalities suggestive of cerebellar dysfunction (saccadic pursuit and/or nystagmus) were present in only 13.5% (15/111) of the cohort. Intellectual deficiency was present in 22 families and progressive intellectual disability was reported for only 17 patients (Table 1). The mean duration was 18 years (18.5 ± 14.7, n = 546, range 0 to 75 years). The cohort was divided into two groups: cases of relatively low duration (below the mean) and cases of relatively long duration (above the mean). The long duration cases more often showed proximal weakness (131/201 versus 66/173; chi-square test, P < 0.0001), greater upper limb reflexes (146/196 versus 108/172; chi-square test, P = 0.02), and lower sense of vibration (133/188 versus 92/158; chi-square test, P = 0.02). Spasticity at rest (159/175 versus 116/143; chi-square test, P = 0.01) was more severe for the long duration group, whereas the prevalence of gait spasticity was similar for the two groups.
Clinical presentation of analysed SPG4-HSP patients
| All patients | Females | Males | P | |
|---|---|---|---|---|
| n | 636 | 302 | 334 | - |
| Mean age at onset, years (n) | 29.3 ± 18.6 (547) | 30.8 ± 18.9 (252) | 28 ± 18.3 (295) | 0.13 |
| Mean age at examination, years (n) | 47.1 ± 17.8 (632) | 48.3 ± 17.5 (302) | 46 ± 18.2 (330) | 0.11 |
| Mean stage of disability (n) | 2.9 ± 1.6 (481) | 2.8 ± 1.6 (225) | 3 ± 1.5 (256) | 0.2 |
| Mean disease duration (n) | 18.5 ± 14.7 (546) | 18.7 ± 16.3 (252) | 18.7 ± 13.4 (294) | 0.47 |
| Gait spasticity, % | 88 (367/417) | 85.2 (161/189) | 90.4 (206/228) | 0.10 |
| Severe, % | 39.8 (146/367) | 37.8 (61/161) | 41.2 (85/206) | 0.52 |
| Spasticity at rest, % | 79.4 (293/369) | 73 (127/174) | 85.1 (166/195) | 0.003 |
| Severe, % | 26.3 (77/293) | 23.6 (30/127) | 28.3 (47/166) | 0.42 |
| Increased lower-limb reflexes, % | 95 (410/431) | 95.5 (193/202) | 94.8 (217/229) | 0.7 |
| Extensor plantar reflex, % | 83.4 (347/416) | 83.5 (157/188) | 83.3 (190/228) | 0.96 |
| Increased upper limb reflexes, % | 65.4 (275/420) | 74.6 (144/193) | 57.7 (131/227) | 0.0002 |
| Hoffman sign, % | 39.7 (95/239) | 47.7 (51/107) | 33.3 (44/132) | 0.02 |
| Proximal lower limb weakness, % | 48.4 (202/417) | 53.8 (99/184) | 44.2 (103/233) | 0.05 |
| Pes cavus, % | 22.4 (86/383) | 24.7 (42/170) | 20.7 (44/213) | 0.34 |
| Decreased sense of vibration at the ankles, % | 61 (241/395) | 66.5 (119/179) | 56.5 (122/216) | 0.04 |
| Urinary symptoms, % | 77 (168/218) | 80.2 (77/96) | 74.6 (91/122) | 0.32 |
| Intellectual impairment, % | 4.2 (17/402) | 5.5 (10/183) | 3.2 (7/219) | 0.26 |
| All patients | Females | Males | P | |
|---|---|---|---|---|
| n | 636 | 302 | 334 | - |
| Mean age at onset, years (n) | 29.3 ± 18.6 (547) | 30.8 ± 18.9 (252) | 28 ± 18.3 (295) | 0.13 |
| Mean age at examination, years (n) | 47.1 ± 17.8 (632) | 48.3 ± 17.5 (302) | 46 ± 18.2 (330) | 0.11 |
| Mean stage of disability (n) | 2.9 ± 1.6 (481) | 2.8 ± 1.6 (225) | 3 ± 1.5 (256) | 0.2 |
| Mean disease duration (n) | 18.5 ± 14.7 (546) | 18.7 ± 16.3 (252) | 18.7 ± 13.4 (294) | 0.47 |
| Gait spasticity, % | 88 (367/417) | 85.2 (161/189) | 90.4 (206/228) | 0.10 |
| Severe, % | 39.8 (146/367) | 37.8 (61/161) | 41.2 (85/206) | 0.52 |
| Spasticity at rest, % | 79.4 (293/369) | 73 (127/174) | 85.1 (166/195) | 0.003 |
| Severe, % | 26.3 (77/293) | 23.6 (30/127) | 28.3 (47/166) | 0.42 |
| Increased lower-limb reflexes, % | 95 (410/431) | 95.5 (193/202) | 94.8 (217/229) | 0.7 |
| Extensor plantar reflex, % | 83.4 (347/416) | 83.5 (157/188) | 83.3 (190/228) | 0.96 |
| Increased upper limb reflexes, % | 65.4 (275/420) | 74.6 (144/193) | 57.7 (131/227) | 0.0002 |
| Hoffman sign, % | 39.7 (95/239) | 47.7 (51/107) | 33.3 (44/132) | 0.02 |
| Proximal lower limb weakness, % | 48.4 (202/417) | 53.8 (99/184) | 44.2 (103/233) | 0.05 |
| Pes cavus, % | 22.4 (86/383) | 24.7 (42/170) | 20.7 (44/213) | 0.34 |
| Decreased sense of vibration at the ankles, % | 61 (241/395) | 66.5 (119/179) | 56.5 (122/216) | 0.04 |
| Urinary symptoms, % | 77 (168/218) | 80.2 (77/96) | 74.6 (91/122) | 0.32 |
| Intellectual impairment, % | 4.2 (17/402) | 5.5 (10/183) | 3.2 (7/219) | 0.26 |
P-values were assessed using a chi-square test.
Clinical presentation of analysed SPG4-HSP patients
| All patients | Females | Males | P | |
|---|---|---|---|---|
| n | 636 | 302 | 334 | - |
| Mean age at onset, years (n) | 29.3 ± 18.6 (547) | 30.8 ± 18.9 (252) | 28 ± 18.3 (295) | 0.13 |
| Mean age at examination, years (n) | 47.1 ± 17.8 (632) | 48.3 ± 17.5 (302) | 46 ± 18.2 (330) | 0.11 |
| Mean stage of disability (n) | 2.9 ± 1.6 (481) | 2.8 ± 1.6 (225) | 3 ± 1.5 (256) | 0.2 |
| Mean disease duration (n) | 18.5 ± 14.7 (546) | 18.7 ± 16.3 (252) | 18.7 ± 13.4 (294) | 0.47 |
| Gait spasticity, % | 88 (367/417) | 85.2 (161/189) | 90.4 (206/228) | 0.10 |
| Severe, % | 39.8 (146/367) | 37.8 (61/161) | 41.2 (85/206) | 0.52 |
| Spasticity at rest, % | 79.4 (293/369) | 73 (127/174) | 85.1 (166/195) | 0.003 |
| Severe, % | 26.3 (77/293) | 23.6 (30/127) | 28.3 (47/166) | 0.42 |
| Increased lower-limb reflexes, % | 95 (410/431) | 95.5 (193/202) | 94.8 (217/229) | 0.7 |
| Extensor plantar reflex, % | 83.4 (347/416) | 83.5 (157/188) | 83.3 (190/228) | 0.96 |
| Increased upper limb reflexes, % | 65.4 (275/420) | 74.6 (144/193) | 57.7 (131/227) | 0.0002 |
| Hoffman sign, % | 39.7 (95/239) | 47.7 (51/107) | 33.3 (44/132) | 0.02 |
| Proximal lower limb weakness, % | 48.4 (202/417) | 53.8 (99/184) | 44.2 (103/233) | 0.05 |
| Pes cavus, % | 22.4 (86/383) | 24.7 (42/170) | 20.7 (44/213) | 0.34 |
| Decreased sense of vibration at the ankles, % | 61 (241/395) | 66.5 (119/179) | 56.5 (122/216) | 0.04 |
| Urinary symptoms, % | 77 (168/218) | 80.2 (77/96) | 74.6 (91/122) | 0.32 |
| Intellectual impairment, % | 4.2 (17/402) | 5.5 (10/183) | 3.2 (7/219) | 0.26 |
| All patients | Females | Males | P | |
|---|---|---|---|---|
| n | 636 | 302 | 334 | - |
| Mean age at onset, years (n) | 29.3 ± 18.6 (547) | 30.8 ± 18.9 (252) | 28 ± 18.3 (295) | 0.13 |
| Mean age at examination, years (n) | 47.1 ± 17.8 (632) | 48.3 ± 17.5 (302) | 46 ± 18.2 (330) | 0.11 |
| Mean stage of disability (n) | 2.9 ± 1.6 (481) | 2.8 ± 1.6 (225) | 3 ± 1.5 (256) | 0.2 |
| Mean disease duration (n) | 18.5 ± 14.7 (546) | 18.7 ± 16.3 (252) | 18.7 ± 13.4 (294) | 0.47 |
| Gait spasticity, % | 88 (367/417) | 85.2 (161/189) | 90.4 (206/228) | 0.10 |
| Severe, % | 39.8 (146/367) | 37.8 (61/161) | 41.2 (85/206) | 0.52 |
| Spasticity at rest, % | 79.4 (293/369) | 73 (127/174) | 85.1 (166/195) | 0.003 |
| Severe, % | 26.3 (77/293) | 23.6 (30/127) | 28.3 (47/166) | 0.42 |
| Increased lower-limb reflexes, % | 95 (410/431) | 95.5 (193/202) | 94.8 (217/229) | 0.7 |
| Extensor plantar reflex, % | 83.4 (347/416) | 83.5 (157/188) | 83.3 (190/228) | 0.96 |
| Increased upper limb reflexes, % | 65.4 (275/420) | 74.6 (144/193) | 57.7 (131/227) | 0.0002 |
| Hoffman sign, % | 39.7 (95/239) | 47.7 (51/107) | 33.3 (44/132) | 0.02 |
| Proximal lower limb weakness, % | 48.4 (202/417) | 53.8 (99/184) | 44.2 (103/233) | 0.05 |
| Pes cavus, % | 22.4 (86/383) | 24.7 (42/170) | 20.7 (44/213) | 0.34 |
| Decreased sense of vibration at the ankles, % | 61 (241/395) | 66.5 (119/179) | 56.5 (122/216) | 0.04 |
| Urinary symptoms, % | 77 (168/218) | 80.2 (77/96) | 74.6 (91/122) | 0.32 |
| Intellectual impairment, % | 4.2 (17/402) | 5.5 (10/183) | 3.2 (7/219) | 0.26 |
P-values were assessed using a chi-square test.
Comparison according to sex
A comparison between males and females revealed no significant differences for age at onset or disorder severity. However, females showed greater upper limb reflexes and a higher frequency of Hoffmann sign than males, despite a similar mean duration of the disorder for both groups (Table 1). Apart from spasticity at rest, females appeared to express a more severe and diffuse disorder phenotype. In contrast, they were more often unable to indicate the age at onset and the proportion of asymptomatic carriers was higher for females than males, suggesting sex-linked penetrance. Penetrance of the disorder was indeed higher in males (0.94, 302/320 versus 0.88, 258/291; chi-square test, P = 0.01) for the entire evolution of the disease (P = 0.01, log-rank test) (Supplementary Fig. 1). The difference in penetrance was even higher when considering only early onset patients, examined before 30 years of age (0.91, 52/57 for males versus 0.7, 35/48 for females; chi-square test, P = 0.02).
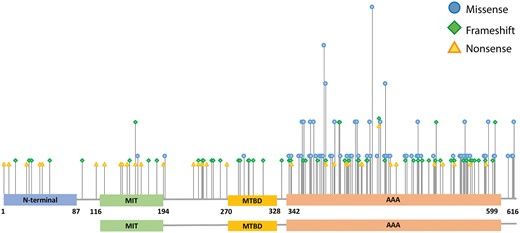
Distribution of missense, nonsense, and frameshift mutations. The height of the bars represents the number of different base changes affecting the same amino acid position of the gene sequence.
Nature and distribution of mutations
We detected a total of 266 different mutations, 134 were previously unreported (Supplementary Table 1). Missense mutations were the most frequent (33%, 87/266), followed by frameshift (24%, 65/266), splice-site (16%, 42/266), and nonsense (13%, 34/266) mutations and deletions (12%, 31/266). Duplications encompassing one or several exons, even involving the 3′ UTR in one patient, and small in-frame amino acid deletions (3%, 7/266) were less frequent. Most (77%, 205/266) pathogenic mutations clustered in the AAA cassette, between amino acids 342 and 599. The observed clustering was particularly evident for missense mutations, predominantly detected in the spastin AAA cassette (96.6%, 84/87) (Figs 2 and 3). Three missense mutations located outside the AAA cassette were classified as possibly pathogenic because they segregated with the disorder in the affected families (Families SAL-055, SAL-1143, SAL-1045). Deletions also tended to cluster in the C-terminus, mostly causing the loss of small portions of the gene, except for five large deletions, which in some cases led to the loss of the entire gene sequence.
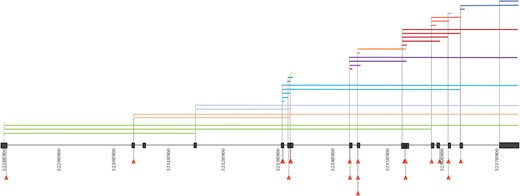
Distribution of deletions and splice-site mutations. SPAST exons are represented in black. Deletions are represented in the upper part of the figure by coloured horizontal bars, spanning the length of the deleted region. Splice-site mutations are represented in the lower part of the figure by red triangles.
We detected a phenocopy in one family (Family SAL-115): a 39-year-old male did not carry the mutation (c.1450_1451del/p.Gly484Cysfs*) responsible for the phenotype in 12 other affected family members. He had experienced vertigo, cramps, and pain from the age of 35. Examination revealed increased brisk tendon reflexes, indifferent plantar reflex, mild scoliosis, and normal MRI and conduction velocities on electromyography.
The GnomAD database was useful for assessing the frequency of the detected mutations in the general population. Only 11 of the 266 identified mutations were present in GnomAD (Supplementary Table 1), all at a frequency <0.1%, providing further evidence in favour of their role in the pathogenicity of SPG4-HSP.
Phenotype–genotype correlation
We analysed the nature of the pathogenic mutation and its position to determine their possible influence on the clinical phenotype. The phenotype observed for mutations leading to a truncated protein (frameshift, nonsense, splice site, and small deletions) were compared to the phenotype of missense mutations. Only mutations affecting spastin after amino acid 342 were considered (n = 505), as most pathogenic mutations altered the AAA cassette.
Individuals with missense mutations showed a much lower mean age at onset (23 ± 19.7, n = 180 versus 33.4 ± 16.7, n = 245; Mann-Whitney test, P < 0.0001) (Fig. 4A). The age at first clinical examination was also lower (41.8 ± 19.6, n = 208 versus 50.2 ± 15.5, n = 285; Mann-Whitney test, P < 0.0001) (Fig. 4B). The distribution of the age at onset was bimodal: the clinical symptoms of most missense-mutation carriers first appeared before the second decade, whereas the first appearance of clinical symptoms of truncating-mutation carriers clustered between the second and sixth decades (Fig. 4C). There was no difference in the severity (3.1 ± 1.7, n = 148 versus 2.8 ± 1.5, n = 224; Mann-Whitney test, P = 0.11) or duration (19.8 ± 15.7, n = 180 versus 17.7 ± 14.2, n = 243; Mann-Whitney test, P = 0.28) of the disorder between missense- and truncating-mutation carriers. However, intellectual disability was significantly more frequent among missense mutation carriers (21.7% versus 4.7%; chi-square test P < 0.0001).
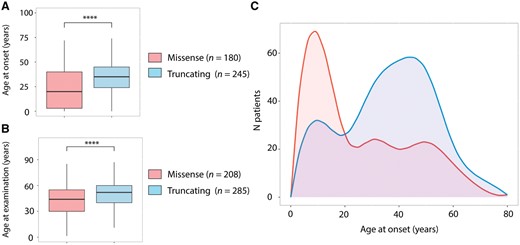
Age at onset distribution and genotype correlations. (A and B) Boxplots representing the age at examination /at onset for patients carrying missense or truncating mutations (Mann-Whitney test, ****P < 0.0001). (C) Distribution of the age at onset of missense- and truncating-mutation carriers. The lower age at onset linked to missense mutations is evident, shown by the density curve (red), characterized by a first peak between birth and the first decade of life and a second smaller peak between the third and fifth decades. Truncating-mutation carriers (blue curve) are characterized by a later age at onset, with a small peak between birth and the first decade of life and a major peak between the second and fifth decades.
Factors influencing the disorder severity and progression
We assessed whether the severity of the disorder was influenced by the age at onset by dividing the cohort into early onset cases (below the median age of onset of 30 years) and late onset cases (above the median age of onset) (n = 546) and comparing the mean stage of disability at the latest examination. The disability for late onset cases was more severe than that for early onset cases. This was especially true when the duration of the disorder was between 11 and 30 years (3.2 ± 1.16, n = 86 versus 3.8 ± 0.9, n = 88; Mann-Whitney test, P < 0.0001) (Supplementary Fig. 2).
We performed a more comprehensive analysis of the progression of SPG4-HSP for 116 patients for whom several follow-up examinations were available. Patients with a slow course had a less severe outcome (4.2 ± 1.3, n = 61 versus 3.3 ± 1.5, n = 54; Mann-Whitney test, P = 0.001), which was not explained by their age at onset (slow course group 25.8 ± 17.4, n = 50 versus fast course group 27.4 ± 16.4, n = 60 Mann-Whitney test, P = 0.6) or genotypes (32% missense versus 68% truncating, P = 0.7). Patients with a more rapidly evolving disorder had a higher frequency of urinary incontinence and lower limb proximal weakness (82% versus 48%, P = 0.01 and 64.7% versus 34.5%, P = 0.01), and were more severely affected, as reflected by their disability scores.
The c.131C>T/p.(Ser44Leu) polymorphism
Eleven patients carried the SPAST exon 1 variant c.131C>T/p.(Ser44Leu) associated with a major pathogenic mutation. The S44L variant was associated with a significantly lower age at onset (11 ± 16.9, n = 11 versus 29.3 ± 18.6, n = 547; Mann-Whitney test, P = 0.004) and thus a lower age at the first examination (32 ± 22.2, n = 11 versus 47.1 ± 17.9, n = 632; Mann-Whitney test, P = 0.02). There was no difference in the severity of the disorder for patients with the S44L and those without (2.9 ± 1.6, n = 481 versus 3.3 ± 0.94, n = 10, Mann-Whitney test, P = 0.46). Patients with p.(Ser44Leu) and another SPAST mutation showed pure spastic paraplegia with increased tendon reflexes, a reduced sense of vibration at the ankles, a bilateral extension plantar response, and urinary urgency or incontinence. The phenotype was more complex in only two cases and accompanied by delayed psychomotor development and moderate or severe sphincter disturbance, probably due to the presence of a missense mutation and early onset (see above genotype-phenotype correlation). In one case, cerebral MRI revealed mild cerebellar atrophy.
Neuropathological examination
We were able to perform a post-mortem neuropathological examination of Case FSP-625-011 (with pre-mortem consent of the patient). This male patient died from cerebral metastases of vesical cancer at 59 years of age. He had been afflicted with spastic paraplegia since the age of 25 years. He inherited the disorder from his mother, was the youngest of three brothers, and the only affected sibling. A SPAST frameshift mutation (c.1215_1219del/p.Asn405Lysfs*36) had been identified and segregated in the family. The stiffness of his legs progressed over the years and he needed a walking aid at 37, two canes at 42, and a wheelchair at 48 years of age. Evaluation by the Spastic Paraplegia Rating Scale (SPRS) gave a score of 43/52 at 55 and 49/52 at 58 years of age. The spasticity of the lower limbs was severe, with weakness and extensor plantar reflexes. Pyramidal signs were marked in the upper limbs. He had a bilateral Hoffmann sign. Mild urinary urgencies were noted since the age of 49. The sense of vibration disappeared at age 43 years of age. Eye gaze and the fundus were normal. No cerebellar or extrapyramidal signs were noted. Brain and medullar MRI were normal at 38 and 55 years of age. Conduction velocities were normal at the age of 59, except for left carpal compression signs. Haematuria led to the discovery of vesical cancer. The patient died 1 year later at the age of 59 due to brainstem compression caused by cerebral haemorrhage.
The brain weighed 1664 g. The right hemi-brain was examined after formalin fixation and the left cryoprotected and sampled immediately post-mortem. Macroscopically, a temporal herniation had caused compression and haemorrhage of the midbrain. There was a large dilation of the lateral ventricles. We observed two metastases of 0.5 cm in diameter. The first was located in the frontobasal region and the second in the vermis of the cerebellum. Microscopically, the number of Betz cells in the motor cortex was low and there were a few neurofilament-positive–MBP-negative spheroids in the white matter; the cerebral cortex was otherwise normal. The thalamus, striatum, cerebellum, and dentate nucleus were normal. The pyramids were small at the level of the medulla oblongata. The posterior part of the lateral column of the spinal cord was pale in a region corresponding to the pyramidal tract. Such myelin pallor was also marked in the dorsal spinocerebellar tract. The cuneiform fasciculus in the posterior column of the spinal cord was also pale. MBP–neurofilament immunohistochemistry showed no demyelination, but degeneration of the affected tracts. We observed axon spheroids and abnormally large fibres with a sausage-like aspect and thin myelin layer in the pyramidal tract (Fig. 5A and B). Remarkably, a high density of very small fibres with thin myelin fibres was clearly shown by MBP-NF double immunohistochemistry on sagittal sections, suggesting regeneration of the corticospinal tract. Tau staining did not show the presence of tau-positive cells in any of the analysed regions. There were no amyloid-β deposits. The pars compacta of the substantia nigra and locus coeruleus showed mild neuronal loss. There were α-synuclein positive fibres in the dorsal nucleus of the vagus nerve and the locus coeruleus, substantia nigra, and amydala. A diagnosis of mild brainstem-type Lewy body disease was made.
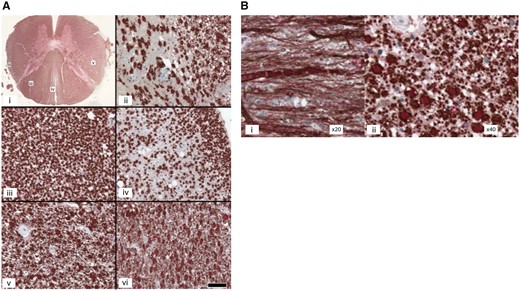
Spinal cord of a SPAST mutated patient. (A) Spinal cord of Patient FSP-625-011: alteration of the spinocerebellar and spinocortical tracts and posterior columns. Double labelling of myelin basic protein/neurofilaments (MBP/NF). (i) Transversal section of the spinal cord at the thoracic level. (ii–vi) Scale bar = 50 µm, MBP: brown/NF: red. (ii) Posterior spinocerebellar tract: loss of myelinated and unmyelinated fibres in the dorsal spinocerebellar tract. (iii) Fasciculus cuneiformis: sparing of the fasciculus cuneiformis. (iv) Fasciculus gracilis: loss of myelinated and unmyelinated fibres. (v) Cortico-spinal lateral tract at the thoracic level: numerous processes of small diameter among unusually large myelinated fibres, suggesting marked regeneration of the corticospinal tract. Arrows indicate clusters of small fibres. (vi) Lateral corticospinal tract at the level of the medulla oblungata: the number of small regenerating fibres appear to be as large in the medulla as at the thoracic level. (B) Pyramidal tract of the spinal cord. (i) Atypically large fibres with a sausage-like aspect and a poor myelin layer in longitudinal sections. (ii) Neurodegeneration and axon spheroids detected in transversal sections.
The metastases had the characteristics of a urothelial carcinoma.
Discussion
We assembled the largest SPG4-HSP cohort (n = 842) to date, which allowed us to show that both the sex of the individual and the nature of the SPAST mutation act as modifiers of the HSP phenotype. The principal aim of this study was to search for factors that can account for the extremely high clinical variability of this disease, even when the same pathological variant is shared (Santorelli et al., 2000). The age at onset ranged from early childhood to the seventh decade and followed a bimodal distribution, with the first peak before the first decade of life and the second lower peak between the second and fifth decades, similar to that observed in a Dutch SPG4 cohort (de Bot et al., 2010). The underlying cause of such a bimodal distribution was missing from previous reports. Here, we show that the mutation type may explain the bimodal distribution, with the first-decade peak mainly due to missense mutations, whereas the second is most likely due to truncating mutations. However, it is important to underline the difficulty of assessing the precise age at onset, leading to a possible bias due to the wrong age or to the fact that early-onset patients are examined earlier. In addition, we could not include individuals for whom age at onset information was not available. Yet, the distribution of missense versus truncating mutations in this cohort was similar (30% versus 70%, n = 206 compared to 33% versus 65%, n = 636).
Among the 266 different mutations that we report here, most were located in the spastin AAA domain, in accordance with previous reports (Fonknechten et al., 2000; Lindsey et al., 2000; Svenson et al., 2001; Meijer et al., 2002; Charvin et al., 2003; Shoukier et al., 2009; Alvarez et al., 2010; Loureiro et al., 2013). Truncating mutations, with the loss of function of spastin as a possible consequence, comprised most of the mutations. Missense mutations clustered in the AAA cassette, whereas pathogenic frameshift and nonsense mutations were distributed throughout the gene. Missense- and truncating-mutation carriers showed no difference in disorder severity based on evaluation using a disability scale. Nevertheless, a notable exception was intellectual disability, which was more frequent among missense carriers. Case reports of families that were included in our cohort had already suggested this possible association (Ribaï et al., 2008). In addition, we previously observed that missense-mutation carriers show greater cognitive decline (Tallaksen et al., 2003).
In our cohort, we observed, for the first time, a correlation between the presence of a missense mutation that affects the AAA domain and an earlier age at onset when comparing missense and truncating mutation carriers. No clear genotype–phenotype correlation has been previously established for the possible association between the nature of the mutation and age of onset or severity of the disorder. Indeed, prior attempts have led to different conclusions (Fonknechten et al., 2000; Svenson et al., 2001; Proukakis et al., 2003; Patrono et al., 2005; Depienne et al., 2007; Shoukier et al., 2009; de Bot et al., 2010; Loureiro et al., 2013), possibly due to small sample size.
There is an ongoing scientific debate concerning the pathogenic mechanisms underlying the age at onset depending on the type of SPAST mutation. Loss of function and consequent haploinsufficiency have been the most plausible pathogenic mechanisms to explain SPG4-HSP onset, as truncating mutations are the most frequent cause of SPG4-HSP (Fonknechten et al., 2000; Sauter et al., 2002; Beetz et al., 2006; Depienne et al., 2007; Shoukier et al., 2009; Alvarez et al., 2010) and most are predicted to predominantly affect the enzymatic activity of the spastin domain, thus reducing its ATPase activity (Bürger et al., 2000; Fonknechten et al., 2000; Lindsey et al., 2000; Molon et al., 2004; Beetz et al., 2006; Depienne et al., 2007). However, missense mutations that affect the AAA domain were shown to lead to constitutive binding of spastin to microtubules, suggesting a dominant-negative mechanism (Errico et al., 2002). Moreover, it was shown that missense mutations outside the AAA domain lead the M1 isoform of spastin to negatively interact with spastin-M87, diminishing its microtubule-severing activity (Solowska et al., 2010). The observation of an earlier age at onset for missense carriers supports a loss-of-function mechanism, but does not exclude the possibility that a dominant-negative mechanism could be associated with specific missense mutations. Among the analysed cohort, this could be true for those patients (30%, 17/56) with an onset before 20 years of age who carry missense mutations already known to have a possible dominant-negative effect (Errico et al., 2002). We were unable to link the severity of the disorder to the nature or position of the mutation, but confirmed that later onset was linked to faster progression (Fonknechten et al., 2000). Multiple follow-up analyses showed that proximal weakness of the lower limbs and urinary incontinence were the symptoms most frequently associated with rapidly evolving disease.
The sex of the individual appeared to be a second factor that influences the age at onset in our cohort. Until now, no clear sex-related effect has been reported (Schüle et al., 2016), but a higher prevalence in males has already been observed (Proukakis et al., 2011). Females of our cohort with a similar duration of the disorder had a more severe and diffuse form of the disorder, characterized by upper limb involvement. In contrast, the number of asymptomatic females was higher than asymptomatic males, leading to a lower penetrance estimate for females, especially before the third decade. It is possible that neurosteroid progesterone and oestrogens could protect females (Orlacchio et al., 2005; Proukakis et al., 2011). Based on our observations, protective factors could delay onset of the disorder, but once started, its evolution is more rapid and severe than in males. This is comparable to sex-linked differences observed in Alzheimer’s disease, with affected females showing longer survival but a more severe disability (Sinforiani et al., 2010).
The most extreme aspect of phenotypic variability was incomplete penetrance, i.e. individuals that do not express the disorder even though they carry the SPAST mutation. Fifty-one (8.3%) carriers declared themselves to be asymptomatic but only 23 were truly asymptomatic when examined. Overall penetrance was estimated to be 0.9, with half of the patients manifesting the disorder at 45 years of age and complete penetrance by the seventh decade. A highly significant correlation for the age at onset between sibling and parent-offspring pairs was less variable than between different families. The highest correlation was observed for same-sex sib-pairs, especially between brothers. This suggests that genetic factors may have a major impact on the age at onset. A sex-linked effect may also come into play, enhancing the correlation between siblings for the age at onset.
The phenotype in the overall cohort did not differ from the already reported features typical of SPG4-HSP (Durr et al., 1993): a pure form of spastic paraplegia with evident but mildly severe spasticity at gait and proximal weakness, as well as increased upper limb reflexes in 65% of patients. Many studies tried to link additional phenotypical presentations to the core spastic phenotype such as dementia, intellectual deficiency, neuropathy, tremor but also Dandy Walker malformation in one family and one sporadic case (Durr et al., 1993; Tallaksen et al., 2003; McDermott et al., 2006; Ribaï et al., 2008; Scuderi et al., 2008; van de Warrenburg, 2008; Murphy et al., 2009; Parodi et al., 2017). In the latter, the malformation could be linked to the SPAST mutation in the described family, but also to another, yet unknown mutated gene that is in linkage disequilibrium with SPG4 or a modifier variant (Scuderi et al., 2008; van de Warrenburg, 2008). Neuropathy and tremor are frequent neurological signs, and it is difficult to prove the link to SPAST variants and could be considered as coincidental. Subtle cognitive impairment was associated with SPG4, especially linked to missense variants (Tallaksen et al., 2003) but without evidence in rare neuropathological cases reported to date (see below).
A known modifier, the SPAST c.131C>T/p.(Ser44Leu) variant (S44L) in exon 1, was present in 11 patients, in addition to a causative pathogenic SPAST mutation. As expected, the age at onset was significantly earlier, as S44L was associated with a SPAST mutation, but the disorder was neither more severe nor associated with additional neurological features in this group of patients. These results confirm that S44L modifies the age at onset (Svenson et al., 2004; McDermott et al., 2006). S44L affects only the M1 spastin isoform. It has thus been proposed that S44L might act in a dominant-negative fashion on the M1 isoform, lowering the microtubule-severing activity of the M87 isoform (Solowska et al., 2010). Further studies of the genetic modifiers of the evolution of the disorder are warranted.
Neuropathological examination showed myelin pallor in the pyramidal tract and dorsal column of the spinal cord. This was due to degeneration of the tracts (axons and myelin were affected) rather than demyelination. There were changes in the white matter of the motor cortex, indicating that the entire length of the pyramidal tract was affected. However, a number of Betz cells were still present, suggesting that the defect did not primarily involve the cell body. In contrast, the lesions predominated in the spinal cord, in agreement with the possibility of a dying-back mechanism. The small size of the pyramids (at the level of the medulla oblongata), raise the possibility of developmental hypoplasia preceding atrophy. The large number of thin axons in the pyramidal tracts suggests a regenerating process, although it is not currently possible to discard the alternative hypothesis of primary atrophy of the axons.
We did not find the tauopathy or amyloid-β accumulation reported in another SPG4-HSP post-mortem analysis (Wharton et al., 2003).
In conclusion, pure loss-of-function mutations (truncating mutations) are associated with a later onset than missense mutations, which probably result in a dominant-negative effect in addition to the loss of function. Sex-linked factors could be protective in female carriers, leading to lower penetrance. Genetic modifiers are clearly implicated in such variability, since intra-familial correlations were stronger within nuclear families, particularly between siblings of the same sex. Further molecular studies should allow the identification of genes that modify the age at onset other than the p.(Ser44Leu) variant, for which effect was confirmed in this study.
Abbreviations
Acknowledgements
Many thanks to patients for their participation and to all the SPATAX network collaborators (see Appendix 1).
Funding
The present work was funded by the VERUM Foundation for Behaviour and Environment (project: MODIFSPA).
Competing interests
The authors report no competing interests.
Appendix 1
SPATAX network collaborators
Myriem Abada, Mathieu Anheim, Dominique Bonneau, Perrine Charles, Pierre Clavelou, Giulia Coarelli, Paula Coutinho, Rabab Debs, Nizard Elleuch, Claire Ewenczyk, Imed Feki, Xavier Ferrer, Bertrand Fontaine, Cyril Goizet, Lucie Guyant-Marechal, Didier Hannequin, Solveig Heide, Abdoul Kassar, Pierre Labauge, Alain Lagueny, Isabelle Le Ber, Thomas Lenglet, Lionel Maldergem, Cecilia Marelli, Karine Nguyen, Diana Rodriguez, Tanya Stojkovic, Alina Tataru, Maya Tchikviladze, Christine Tranchant, and Nadia Vandenberghe.
References
Author notes
Appendix 1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}