




This scientific commentary refers to ‘Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic-rescue by TMEM106B knockdown’, by Clayton et al. (doi:).
Frontotemporal dementia (FTD) is a neurodegenerative syndrome characterized by atrophy of the frontal and temporal lobes, leading to alterations in behaviour, personality and language. Early symptoms can manifest as mildly as inappropriate comments, word-finding difficulty and apathy towards loved ones, or as more severe changes such as criminal behaviours (e.g. theft, sexual disinhibition) and reckless financial decisions, which can tragically lead to alienation of friends and family. FTD is one of the leading causes of early-onset dementia and is the third most common cause of dementia overall. Although historically observed to co-occur with amyotrophic lateral sclerosis (ALS), recent studies showing overlap of several genetic and neuropathological features have helped to reframe FTD and ALS as existing on a spectrum with features of motor neuron disease in up to 40% of patients with FTD. Mutations in the genes C9orf72, MAPT and GRN account for about 60% of inherited FTD cases, while rarer mutations occur in other genes such as VCP, TBK1, CHMP2B, TARDBP and FUS (Bang et al., 2015). Although rare, mutations in these genes have helped to illuminate key cellular pathways that might lead to novel therapeutic strategies. Recent work has converged on the endolysosomal system, the cell’s local ‘trash sorting and recycling plant’. Cellular components that need to be recycled are packaged into the appropriate shipping containers (endosomes) and sent to the garbage disposal (lysosomes). Lysosomes contain degradative enzymes that break down the material into smaller bits, which can then be reused by the cell. Mutations in GRN and CHMP2B both lead to lysosomal impairments, and the FTD risk gene TMEM106B is crucial for trafficking of lysosomes within dendrites (Schwenk et al., 2014; Clayton et al., 2015). Compellingly, recent work found that the C9orf72 gene, mutation of which is the most common genetic cause of both FTD and ALS, is also necessary for normal endolysosomal function. Chemical and genetic modulation of this system rescued motor neuron degeneration in cells harbouring C9orf72 mutations (Shi et al., 2018). In this issue of Brain, Clayton and co-workers add to the evidence linking endolysosomal dysfunction to FTD by demonstrating that an FTD-causing mutation in CHMP2B leads to defects in the ability of endosomes to properly traffic within dendrites, resulting in abnormal dendritic branching. Importantly, they were able to rescue these phenotypes with a therapeutic strategy targeting TMEM106B (Clayton et al., 2018).
FTD-causing mutations in CHMP2B produce truncated versions of CHarged Multivesicular Protein 2B, a subunit of a larger complex (ESCRT-III) responsible for membrane cleavage during endolysosomal trafficking. The authors have previously described a transgenic mouse line that expresses a mutated form of CHMP2B found in patients with FTD (CHMP2BIntron5) (Ghazi-Noori et al., 2012). In the current study, they analysed primary cortical neurons from these mice for cellular defects. They found that neurons displayed a reduced number of endolysosomes in their cell bodies when expressing this mutant protein at physiological levels (Fig. 1). They next asked whether this diminished population could be the result of defects in the trafficking of the endolysosomes. They turned to live cell imaging and found a striking reduction in the motility of these vesicles. Remembering that the ESCRT complex needs to be separated from the endolysosomal membrane for efficient trafficking, Clayton et al. hypothesized that the mutant CHMP2B was getting stuck on the membrane and that this was responsible for the observed stalling. They used a clever technique to test this hypothesis. After expressing a fluorescently-tagged form of CHMP2B, they bleached a part of the cell with a focused laser and measured how long it took for the fluorescence to recover. In wild-type neurons, the recovery was rapid, indicating that the CHMP2B proteins could be readily exchanged from the membrane, bleached for unbleached. However, the fluorescence did not recover in the neurons expressing mutant CHMP2B, consistent with it being stuck on the membrane. The authors were able to offer a possible explanation for this finding by showing that mutant CHMP2B was unable to recruit the protein VPS4, an enzyme critical for separating the ESCRT complex from the endolysosomal membrane.
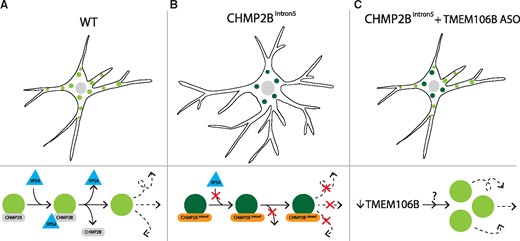
Reducing levels of TMEM106B rescues the endolysosomal trafficking block and dendritic arborization abnormalities in primary cortical neurons expressing mutant CHMP2B. (A) Wild-type (WT) neurons require recruitment of the protein VPS4 to help detach CHMP2B from the membrane to allow efficient endolysosomal trafficking. (B) Neurons expressing mutant CHMP2B (CHMP2BIntron5) have increased dendritic branching with a reduced number of endolysosomes within the cell body, likely owing to the inability of mutant CHMP2B to recruit VPS4, rendering these vesicles stationary. (C) Treatment with an antisense oligonucleotide (ASO) against TMEM106B rescues the increase in dendritic branching and broadly increases trafficking without mobilizing endolysosomes carrying mutant CHMP2B.
The authors next asked how this striking endolysosomal defect could lead to impairments in neuronal function. They found that the dendrites of neurons expressing mutant CHMP2B had significantly more branches than those of wild-type neurons. This dendritic branching phenotype is reminiscent of TMEM106B, but with opposite effects. That is, in a previous study knockdown of TMEM106B resulted in increased lysosomal transport with decreased dendritic outgrowth (Schwenk et al., 2014), whereas in the current study Clayton and co-workers observed decreased lysosomal transport and increased dendritic branching. Because of these opposing effects, the authors reasoned that they might be able to rescue the mutant CHMP2B defects by reducing the levels of TMEM106B.
One way to reduce the level of a protein is to target the mRNA that encodes it. Antisense oligonucleotides (ASOs) are highly stable, synthetic nucleic acids that can target specific mRNA sequences for degradation. They have emerged as a promising therapeutic strategy for neurodegenerative diseases, with translational success most notable in the US Food and Drug Administration approval of nusinersen for patients with spinal muscular atrophy. ASO therapy has demonstrated dramatic rescue in preclinical studies of TDP-43 and C9orf72 mouse models of ALS with promising results in models of other expanded nucleotide repeat diseases such as myotonic dystrophy, Huntington’s disease and spinocerebellar ataxia type 2 (Scoles et al., 2017). Clayton et al. tested ASOs targeting TMEM106B in primary neuronal cultures from mice expressing mutant CHMP2B. This treatment rescued the increase in dendritic arborization and decreased mobility of endolysosomal structures (Fig. 1). Thus, lowering levels of TMEM106B is sufficient to rescue the phenotypes of neurons expressing mutant CHMP2B. The rescue did not result from mobilizing the mutant CHMP2B structures or enhancing their ability to recruit VPS4, but instead entailed increasing trafficking more broadly, perhaps by mobilizing previously quiescent vesicles.
One of the challenges facing ASO therapy is the question of whether a single drug can be broadly applied to a clinical disease with myriad causative genetic mutations. The CHMP2B FTD-causing mutations are rare (<1%) among inherited FTD cases (Bang et al., 2015) and an ASO targeting CHMP2B might have therapeutic benefit only within patients harbouring these mutations. However, Clayton et al. show that the defects observed in this study can be rescued by an ASO against TMEM106B, one of the most widely recognized risk factors for FTD (Van Deerlin et al., 2010). Previous work identified TMEM106B as part of a molecular brake in the endolysosomal pathway (Schwenk et al., 2014). This supports the therapeutic strategy of reducing TMEM106B to broadly increase trafficking to compensate for arrested endolysosomes that may occur secondary to several genetic mutations. Indeed, recent work found that deletion of TMEM106B from GRN knockout mice rescued an increase in lysosomal enzymes as well as an increase in motor activity and disinhibition (demonstrated by more frequent exploration of the open areas of a maze) (Klein et al., 2017). However, other researchers found that breeding mice missing one copy of Grn with mice missing one copy of Tmem106b did not rescue most lysosomal abnormalities and socialization deficits observed in the GRN haploinsufficient mice (Arrant et al., 2018).
The work of Clayton et al. supports the hypothesis that endolysosomal dysfunction contributes to FTD and highlights a potential therapeutic strategy. However, some questions remain. First, ASO-mediated reduction of TMEM106B in vivo would not be specific to damaged and dying neurons. Clayton et al. found that the ASOs did not have a strong effect on trafficking or branching of wild-type neurons. However, previous reduction of TMEM106B using shRNA (causing stronger knockdown than with ASOs) in rat hippocampal neurons showed that TMEM106B was essential for lysosomal localization and trafficking as well as for the creation and maintenance of a complex dendritic arbour (Schwenk et al., 2014). Clayton et al. posit that perhaps their milder reduction was insufficient to affect healthy cells but effective in mutation-carrying neurons, raising the possibility that there may be an optimal targeting dosage that does not cripple healthy cells.
The larger question, of course, is how well will these findings translate to humans living with FTD? The mutant CHMP2B mouse model does mimic the neuropathological features seen in patients with p62- and ubiquitin-positive inclusions, axonal degeneration and age-related gliosis (Ghazi-Noori et al., 2012). However, the mice do not have a behavioural phenotype that matches the profound features seen in FTD and this limitation precluded the authors from being able to demonstrate behavioural rescue of TMEM106B knockdown in vivo. Moreover, the longitudinal use of a TMEM106B ASO must be evaluated to examine the dosing frequency required for impaired neurons as well as the long-term effects of reducing TMEM106B in healthy neurons. However, for a genetically heterogeneous syndrome such as FTD, TMEM106B stands as a hopeful target for mobilizing a crippled endolysosomal system and halting the progression of a socially isolating and devastating disease.
Competing interests
The authors report no competing interests.


{kind=link}