




Sir,
We read with interest the article by Ladislau et al. (2018), highlighting the key role of type I interferon (IFN) in the pathophysiology of the muscle disease observed in dermatomyositis and reporting the short-term efficacy of ruxolitinib in four adult patients. Type I IFNs bind a unique common receptor (IFNAR) and subsequently activate the Janus Kinase 1 (JAK1) and tyrosine kinase 2, which, in turn, leads to the phosphorylation of signal transducer of activator of transcription (STAT) 1 and STAT2 driving the expression of IFN-stimulated genes (ISGs). Here, we describe the use of the JAK1/2 selective inhibitor ruxolitinib in a child with severe vasculopathic refractory juvenile dermatomyositis (JDM). Informed parental consent was obtained for the use of ruxolitinib on a compassionate basis and the associated experimental work, conducted in compliance with the 1975 Declaration of Helsinki.
A previously healthy 13-year-old female presented with acute, predominantly proximal muscle weakness, diffuse moderate erythema, and nail bed and palate telangiectasia. At diagnosis the childhood myositis assessment scale (CMAS) was 2/52, the manual muscle testing (MMT) score 38/80 and the disease activity score (DAS) skin 6/9. Creatine kinase was elevated at 12 900 UI/l and anti-NXP2 antibody testing was positive. Deltoid muscle biopsy showed acute and severe ischaemic features as assessed by high total and vascular domain scores. Microinfarcts, obvious capillary loss, microthrombosis and ischaemic punched-out vacuoles were prominent features, associated with perivascular inflammation. Major histocompatibility complex class I antigens (HLA ABC) were diffusely re-expressed by myofibres, with perifascicular reinforcement. Despite high dose intravenous prednisone (1.2 mg/kg/day) and subcutaneous methotrexate (MTX), her condition rapidly worsened. Three weeks after diagnosis she was admitted to the intensive care unit with tetraparesis, dysphonia, dysphagia, ileus, abdominal pain, bloody diarrhoea and worsening cutaneous involvement. She received prednisone (1.6 mg/kg/day) and intravenous immune globulins (IVIG) (2 g/kg for three consecutive days), followed by plasma exchange (four sessions per week) and rituximab (three weekly doses at 375 mg/m2). MTX was discontinued. This regimen resulted in clinical improvement (CMAS: 45/52; MMT: 79/80), so that prednisone was gradually tapered, and plasma exchanges spaced out and eventually discontinued 6 months after diagnosis. However, 3 weeks later, while the patient was maintained on 0.3 mg/kg/day prednisone, muscle weakness and cutaneous lesions recurred (Fig. 1). Consequently, prednisone was increased (0.9 mg/kg/day), IVIG (2 g/kg) given on two consecutive days, and mycophenolate mofetil (MMF) initiated. Additionally, plasma exchanges were recommenced and a fourth dose of rituximab given. Two further muscular, cutaneous and/or gastrointestinal relapses occurred, 12 and 18 months after diagnosis, as prednisone and the frequency of plasma exchange were being tapered. Furthermore, she developed lower limb oedema resulting from diffuse fascia calcinosis.
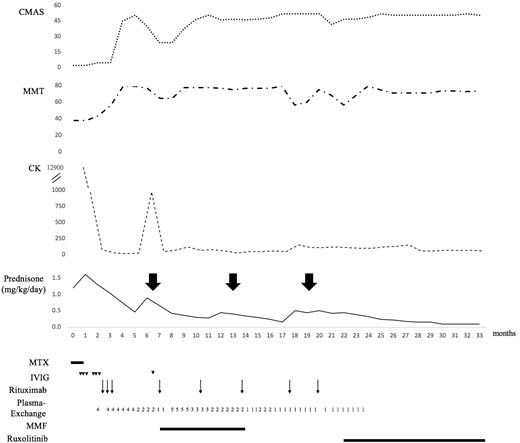
Disease course and treatment regimen. Initially, IVIG was administered on three consecutive days, 2 weeks apart. Arrowheads represent the time points of IVIG dosing (2 g/day). Rituximab was given at a dose of 375 mg/m2/week over three consecutive weeks, and then every 3 to 4 months (arrows). Plasma exchanges were initially performed four times per week, then gradually tapered to once a week and discontinued. They were restarted following a disease flare 7 months after diagnosis. The numbers represent the frequency of plasma exchanges per week. The large bold arrows indicate disease flares. CK = creatine kinase; CMAS = childhood myositis assessment scale; IVIG = intravenous immune globulins; MMF = mycophenolate mofetil; MMT = manual muscle testing; MTX = methotrexate.
Considering the severity of the phenotype and the dependence on prednisone (dosage≥0.5 mg/kg/day) and repeated plasma exchanges, we investigated in our patient the type I IFN pathway in order to consider targeted therapy. We observed an activation of both type I IFN production and signalling (Fig. 2). Specifically, using an ultrasensitive digital ELISA assay (Rodero et al., 2017), we recorded higher serum levels of IFN protein, increased expression of IFN-stimulated genes (ISGs) and constitutive phosphorylation of STAT1 and STAT3 in T lymphocytes and monocytes compared with controls (Fig. 2). This constitutive activation of the type I IFN pathway led us to initiate treatment with the JAK1/2 selective inhibitor ruxolitinib (10 mg twice a day, i.e. 0.5 mg/kg/day) 21 months after the diagnosis. Rituximab and MMF were discontinued (Fig. 1). Within the first 2 months of treatment with ruxolitinib, MMT and CMAS scores increased from 47 to 52 and from 57 to 79, respectively. Furthermore, the patient global disease activity score on a 10 cm visual analogue scale (VAS) decreased from 5/10 to 2/10, and physician global disease activity score fell from 5/10 to 1/10 on VAS. Skin disease was moderate, and DAS skin decreased from 4/9 to 0/9. The prednisone dose was progressively decreased from 0.5 mg/kg/day and plasma exchanges were discontinued. At last follow-up, 12 months post-initiation of ruxolitinib, JDM was clinically inactive according to PRINTO criteria and prednisone had been tapered to 0.1 mg/kg/day. Anti-NXP2 antibodies remained detectable. During the 10 months of treatment with ruxolitinib, her height increased from 149 to 151 cm and she had her menarche. Ruxolitinib was well tolerated, without any of the haematological or infectious side effects previously described in adults treated with ruxolitinib for myelofibrosis. Ruxolitinib pharmacokinetics were assessed regularly and were consistent with our previous report (Frémond et al., 2016). Of note, serum levels of IFNα protein remained elevated under treatment (Fig. 2E), as did the IFN score measured on two occasions, 2.5 and 7 months after starting ruxolitinib. However, the marked clinical improvement that we observed was mirrored by an ex vivo flow cytometry assay performed the day of the initiation of ruxolitinib. Blood was collected before the first drug intake (H0) and 4 h (H4) after dosing. At H4, STAT1 constitutive phosphorylation in CD3+ lymphocytes and monocytes was decreased, comparable to a healthy control (Fig. 2F). When the same experiment was repeated 2 months after initiation of ruxolitinib, we observed constitutive phosphorylation of STAT1 at H0 only in monocytes (data not shown).
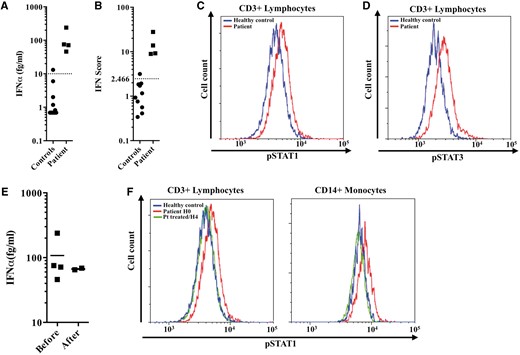
Constitutive activation of the type I interferon pathway and the ex vivo effect of ruxolitinib on constitutive phosphorylation of STAT1. (A) IFNα protein concentration (healthy controls <10 fg/ml) measured in the serum from the patient before treatment with ruxolitinib. (B) IFN score (normal <2.466) of the patient before treatment with ruxolitinib reflecting increased expression of IFN-induced gene transcripts. (C and D) Increased constitutive phosphorylation of STAT1 (C) and STAT3 (D) in CD3+ lymphocytes from the patient compared to a healthy control. (E) IFNα protein concentration (healthy controls <10 fg/ml) measured in the serum from the patient before and during treatment with ruxolitinib. Black lines depict median values. (F) Peripheral blood mononuclear cells (PBMCs) were obtained from the patient before treatment and 4 h after the first ruxolitinib intake. The treatment is taken twice daily. STAT1 constitutive phosphorylation in CD3+ lymphocytes and monocytes was decreased after treatment, being comparable to a healthy control.
JDM is a rare paediatric-onset idiopathic inflammatory myopathy. It is a heterogeneous disease regarding the association (or lack of association) with myositis-specific autoantibodies and pathological stigmata (Deakin et al., 2016; Gitiaux et al., 2016). We have previously reported that vasculopathy-related clinical and pathological features, as observed in our patient, are associated with severe JDM, necessitating the use of at least three treatment lines, including plasma exchange (Gitiaux et al., 2016). One randomized controlled trial in newly diagnosed JDM patients concluded that the combination of corticosteroids and MTX had the best outcome in terms of efficacy and safety (Ruperto et al., 2016). However, treatment failures were reported in 17/46 patients assigned to this combination (Ruperto et al., 2016). In refractory myositis patients, the multicentre ‘Rituximab in Myositis’ trial evaluated rituximab efficacy in 200 patients, including 48 individuals with JDM (Oddis et al., 2013). Although this trial failed to reach its primary endpoint, an improvement was observed in 83% of adult and juvenile dermatomyositis patients by the end of the trial, with improvement strongly associated with the presence of anti-Jo-1 and anti-Mi-2 antibodies (Aggarwal et al., 2014). These features likely reflect the heterogenous nature of JDM, emphasizing the need for novel targeted therapeutic approaches in severe refractory disease.
An upregulation of type I IFN-stimulated genes and chemokines has previously been noted in peripheral blood of patients with dermatomyositis (Baechler et al., 2007). Consistent with the report of Ladislau et al. in adult patients, we previously demonstrated that myogenic progenitor cells derived from muscle biopsies taken from JDM patients are a cellular source of type I IFN, with IFN levels correlating with disease severity (Gitiaux et al., 2018). These features suggested a potential role for JAK inhibitors in the treatment of JDM as well as of dermatomyositis.
To our knowledge, this is the first report of the successful use of ruxolitinib in a child with refractory JDM. Although the follow-up period after initiation of JAK1/2 inhibition is short, ruxolitinib was the only drug that allowed rapid and sustained cessation of plasma exchange therapy and a progressive tapering of corticosteroids. Given that the last infusion of rituximab was performed 2 months before starting ruxolitinib, and that the efficacy of rituximab was observed 4 months later, the likelihood of an effect of the combined treatment is small. Rather, we consider that ruxolitinib alone was responsible for the improvement in our patient's state since (i) eight previous infusions of rituximab did not elicit a sustained clinical remission; and (ii) the remission under ruxolitinib lasted 14 months after the last infusion of rituximab. Unfortunately, we observed no effect on calcinosis, perhaps suggesting that a type I IFN-independent mechanism mediates this devastating complication. In our patient, constitutive activation of type I IFN production and signalling was observed before treatment with ruxolitinib. Neither the serum IFNα nor the IFN score measured in blood normalized in the 12-month period of treatment. However, on one occasion when measured, STAT1 phosphorylation in CD3+ lymphocytes was comparable to a control after dosing. This is consistent with the observation of a marked clinical improvement under JAK inhibitors in patients with TMEM173-activating mutations, where the inhibition of type I IFN signalling measured in the blood was also incomplete (Frémond et al., 2016). Overall, our findings suggest that JAK inhibition may represent an efficacious and well tolerated therapeutic option in a subset of patients with recalcitrant JDM.
Data availability
The authors confirm that the data supporting the findings of this study are available within the letter.
Acknowledgements
We thank the patient for participating in this study. We thank ImmunoQure AG for sharing antibodies for use in the IFNα Simoa assay.
Funding
Marie-Louise Frémond is supported by the Institut National de la Santé et de la Recherche Médicale (grant no. 000427993). Y.J.C. acknowledges funding from the European Research Council (GA 309449: Fellowship to Y.J.C.), a state subsidy managed by the National Research Agency (ANR, France) under the “Investments for the Future” (ANR-10-IAHU-01), and an ANR grant (CE17001002) to Y.J.C. and D.D.
Competing interests
The authors report no competing interests.
References
Author notes
Florence A. Aeschlimann, Marie-Louise Frémond, Yanick J. Crow and Brigitte Bader-Meunier authors contributed equally to this work.


{kind=link}
{kind=link}