




Abstract
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels control neuronal excitability and their dysfunction has been linked to epileptogenesis but few individuals with neurological disorders related to variants altering HCN channels have been reported so far. In 2014, we described five individuals with epileptic encephalopathy due to de novo HCN1 variants. To delineate HCN1-related disorders and investigate genotype–phenotype correlations further, we assembled a cohort of 33 unpublished patients with novel pathogenic or likely pathogenic variants: 19 probands carrying 14 different de novo mutations and four families with dominantly inherited variants segregating with epilepsy in 14 individuals, but not penetrant in six additional individuals. Sporadic patients had epilepsy with median onset at age 7 months and in 36% the first seizure occurred during a febrile illness. Overall, considering familial and sporadic patients, the predominant phenotypes were mild, including genetic generalized epilepsies and genetic epilepsy with febrile seizures plus (GEFS+) spectrum. About 20% manifested neonatal/infantile onset otherwise unclassified epileptic encephalopathy. The study also included eight patients with variants of unknown significance: one adopted patient had two HCN1 variants, four probands had intellectual disability without seizures, and three individuals had missense variants inherited from an asymptomatic parent. Of the 18 novel pathogenic missense variants identified, 12 were associated with severe phenotypes and clustered within or close to transmembrane domains, while variants segregating with milder phenotypes were located outside transmembrane domains, in the intracellular N- and C-terminal parts of the channel. Five recurrent variants were associated with similar phenotypes. Using whole-cell patch-clamp, we showed that the impact of 12 selected variants ranged from complete loss-of-function to significant shifts in activation kinetics and/or voltage dependence. Functional analysis of three different substitutions altering Gly391 revealed that these variants had different consequences on channel biophysical properties. The Gly391Asp variant, associated with the most severe, neonatal phenotype, also had the most severe impact on channel function. Molecular dynamics simulation on channel structure showed that homotetramers were not conducting ions because the permeation path was blocked by cation(s) strongly complexed to the Asp residue, whereas heterotetramers showed an instantaneous current component possibly linked to deformation of the channel pore. In conclusion, our results considerably expand the clinical spectrum related to HCN1 variants to include common generalized epilepsy phenotypes and further illustrate how HCN1 has a pivotal function in brain development and control of neuronal excitability.
Introduction
Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels constitute a unique class of voltage-gated ion channels comprising four different paralogous subtypes encoded by different genes (HCN1–4) in humans (Ludwig et al., 1998; Santoro et al., 1998). Composed of four homomeric or heteromeric subunits, HCN channels are permeable to potassium and, to a lesser extent, sodium. HCN channels are activated upon hyperpolarization, remain open at negative voltages and their opening is potentiated by binding of cAMP (Biel et al., 2009). They conduct an inward, depolarizing current named If (‘funny’ current) in the heart and Ih (‘hyperpolarization-activated’ current) in the brain (Postea and Biel, 2011; DiFrancesco and DiFrancesco, 2015). The generation of this depolarizing current drives the membrane potential back toward the threshold for calcium and sodium channel activation, thereby allowing action potential firing.
In the brain, HCN channels are involved in multiple regulatory functions including the stabilization of resting membrane potential, the integration of synaptic inputs, the modulation of rhythmic oscillatory activity in individual neurons and neuronal networks, and they are therefore essential for the control of neuronal excitability (Poolos et al., 2002; Poolos, 2012; Benarroch, 2013). All four HCN subtypes are expressed in the mammalian brain but their expression greatly varies from one brain region to the other, HCN1 being the predominant isoform expressed in the cortex (Moosmang et al., 1999; Monteggia et al., 2000). Furthermore, HCN subtypes have distinct electrophysiological properties, HCN1 showing the most positive threshold for activation, the fastest activation kinetics, and the lowest sensitivity to cAMP (Chen et al., 2001).
Increasing evidence over the last decade indicated that long-term deregulation of Ih current properties, due to abnormal HCN channel expression or cellular distribution, are early and probably key events of epileptogenic processes in rodent models with acquired seizures (Noam et al., 2011). Interestingly, both the up- and downregulation of HCN channels have been associated with epileptic activity in different animal models (Noam et al., 2011). An increase of HCN1 channels has also been observed in brain tissue of patients with temporal lobe epilepsy after exeresis (Bender et al., 2003). In 2014, we reported that de novo HCN1 missense variants cause an early infantile epileptic encephalopathy (EIEE) sharing common features with Dravet syndrome (Nava et al., 2014a).
Herein, we report on clinical, electrophysiological, genetic and functional data of novel patients with de novo or inherited HCN1 variants. The cohort includes sporadic and familial patients in whom analysis of clinical manifestations uncovered a large spectrum of phenotypes ranging from the very mild and benign febrile seizures, febrile seizures plus, and genetic generalized epilepsy (GGE) to moderately severe forms of genetic generalized epilepsy with febrile seizure plus (GEFS+), focal or unclassified epilepsy and to catastrophic neonatal or infantile epileptic encephalopathy. Interestingly, the cohort further includes four patients with intellectual disability and autism spectrum disorder without seizures. We also characterized the biophysical properties of mutant channels for 12 of 18 novel variants identified, and investigated possible genotype–phenotype correlations.
Materials and methods
Patients
We collected electroclinical and genetic data of 19 patients carrying de novo variants in the HCN1 gene from highly heterogeneous cohorts of patients with paediatric epilepsies, either refractory or benign and with a supposed genetic aetiology. In addition, we studied four families in which HCN1 variants segregated with epilepsy in at least two affected members. Patients and families were recruited from epilepsy and genetic centres around Europe (Italy, France, Netherlands, Germany, UK, Czech Republic, Portugal, Switzerland and Belgium), Brazil, USA, Canada and Australia. Patients 11 and 36 were recruited though Decipher (ID: 260229 and 357848) (Firth et al., 2009). Collection and analysis of retrospective clinical, EEG, neuropsychological and neuroimaging data were assessed using a specific format filled by the treating specialist aiming to obtain accurate and homogenous information. We classified seizure types and epilepsy/syndromes according the International League Against Epilepsy (ILAE) guidelines (Fisher et al., 2014). We also included eight patients with variants altering HCN1 but classified as variants of unknown significance based on unusual inheritance or phenotype: two patients with intellectual disability but no epilepsy carrying de novo HCN1 variants, two patients with intellectual disability or autism spectrum disorder with a heterozygous deletion encompassing exon 4 of HCN1 identified by microarrays, an adopted child with two heterozygous HCN1 variants, and three probands with HCN1 missense variants inherited from healthy parents. Patient 37 was briefly reported previously (Nava et al., 2014a, b); all other patients are novel.
Genetic studies
Genetic studies were independently performed in diagnostic or research laboratories using next generation sequencing. Details on the methods used to detect the HCN1 variants in each patient are indicated in Supplementary Table 1. Sanger traces confirming variant existence were collected for 25/29 index patients. Sanger sequencing was not performed in Patient 3, for whom variant accuracy was supported by a high sequencing depth (>850×). For the remaining three patients (Patients 10, 13 and 41), we obtained the written report of the genetic tests performed by private laboratories but could not directly access sequence data. Variants identified in Patients 2 and 12 were reported previously (Parrini et al., 2017). The American College of Medical Genetics and Genomics (ACMG) criteria and the Intervar interface (http://wintervar.wglab.org/) were used to filter and classify HCN1 variants (Richards et al., 2015; Li and Wang, 2017). The consequences of HCN1 variants were interpreted on isoform NM_021072.3 using Alamut 2.10 (Interactive Biosoftware, France).
Patch-clamp experiments
The plasmid containing the human HCN1 cDNA differing from the HCN1 reference sequence (NM_021072.3) by a deletion of 3 bp encoding one of the 12 glycine residues starting from residue 63 (known polymorphism) was kindly provided by Dr Juliane Stieber (Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany). The properties of the HCN1 channel with this in-frame variant were comparable to that of HCN1 corresponding to the reference sequence. Ten of the novel identified variants (M153I, M243R, K261E, M305L, G391C, G391D, G391S, I397L, S399P and R590Q) were introduced into this HCN1 cDNA with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All constructs were sequenced to ensure that no additional variants were introduced. CHO-K1 cells transiently co-transfected with 1 µg of plasmid expressing the wild-type or a mutant human HCN1 (hHCN1) channel plus pEGFP [at a ratio of 50(pHCN1):1(pEGFP)], using the NeonTM transfection system (Invitrogen), and whole-cell patch clamp experiments were performed at room temperature 24 h after transfection, as previously described (Nava et al., 2014a). For co-expression experiments, an equal amount of wild-type and mutant constructs (0.5 µg of each plasmid) was transfected in the same conditions.
The biophysical properties of M305L, G391C, G391D, G391S and I397L were further studied in HEK293T cells transiently transfected with wild-type and/or mutant constructs using TurboFect™ transfection reagent (Thermo Fisher). Either 1 µg or 0.5 µg of the HCN1-containing vector (pcDNA3) and 0.3 µg of GFP-containing plasmid (pmaxGFP, AmaxaBiosystems) were transfected. In co-expression studies, 0.5 µg each HCN1 plasmid was co-transfected. Green fluorescent cells were recorded by whole-cell patch-clamp at room temperature 24 h after transfection as described previously (Lolicato et al., 2014).
Finally, the C329S and V414M variants were introduced in pIRES-GFP-hHCN1-WT (GenBank: AF488549.1). CHO cells were transiently transfected with 1.5 µg of wild-type or mutant in pIRES-GFP-hHCN1 plasmid using the jetPRIME reagent (Polyplus Transfection, Euroclone). Whole-cell patch clamp experiments were performed at room temperature 48 h after transfection, as described previously (Gravante et al., 2004).
Data analysis
To analyse current density, the steady state current amplitude was measured at the end of each test potential and normalized to cell capacitance. Activation curves were obtained by fitting the normalized tail currents plotted against test voltages with the Boltzmann equation It = It(max) / (1 + exp(V − V1/2) / k where It is the current amplitude of the tail current recorded for a given prepulse and It(max) is the maximum current amplitude of the tail current, V is the voltage of the prepulse, V1/2 is the half-activation potential and k is the inverse slope factor in mV using OriginPro software (OriginLab, Northampton, MA, USA). All data are presented as mean ± standard error of the mean (SEM).
Molecular dynamic simulations
Residues 94 to 403 of the crystal structure of HCN1 (PDB: 5U6P) (Lee and MacKinnon, 2017) were used as starting point of molecular dynamic simulations. Missing residues were added and in silico point variants were introduced using Modeller 9.19 (Fiser et al., 2000), followed by protonation of titratable sidechains according to their estimated pKa value (PROPKA3) (Olsson et al., 2011). The proteins were embedded into an equilibrated palmitoyloleoylphosphatidyl-cholin (POPC) bilayer consisting of 512 lipids using g_membed (Wolf et al., 2010). K+ ions were placed in the selectivity filter at binding site S4 and in the central cavity (Scav). Finally, the internal cavity was solvated with VOIDOO/FLOOD (Kleywegt and Jones, 1994). The resulting system consisted of the protein, 494 POPC, 41911 TIPS3P water, 126 K+ and 114 Cl− (≈ 0.15 mol/l) for the heterozygous mutation, 441941 TIPS3P water, 122 K+ and 114 Cl− for the wild-type channel and 41970 TIPS3P water, 124 K+ and 114 Cl− for the heterozygous mutation, respectively. Simulations employed the GROMACS software package (2018) (Van Der Spoel et al., 2005; Pronk et al., 2013) and CHARMM36m forcefield (Huang and MacKerell, 2013). The integration time step was set to 2 fs. Van der Waals forces were switched to zero between 0.8 and 1.2 nm. Electrostatic interactions were represented using the fast smooth particle-mesh Ewald (PME) method with a Coulomb cut-off of 1.2 nm (Essman, 1995). Hydrogen bonds were kept constant using the LINCS algorithm (Hess, 1997). The velocity-rescale thermostat (Bussi et al., 2007) with a coupling time constant of 0.1 ps was used to keep the simulation temperature at 298 K. The pressure was kept at 1 bar using the Parrinello-Rahman barostat algorithm (Parrinello, 1981) with a coupling constant of 5 ps. Prior to 100 ns production simulation, the system was energy minimized (5000 steps steepest descend) and equilibrated (10 ns NPT simulation). During equilibration, protein heavy atoms were restrained to their initial position by a force constant of 1000 kJ/mol/nm2.
Data availability
The raw data that support the findings of this study are available from the corresponding authors, upon request.
Results
We collected the data of 33 affected individuals carrying 18 different novel HCN1 missense variants classified as pathogenic or likely pathogenic using the ACMG criteria, all absent from the GnomAD database (Fig. 1 and Supplementary Table 1). Overall, 14 different variants occurred de novo in 19 sporadic patients (Table 1). Four variants segregated with epilepsy in at least two affected family members of four families (Table 2). All 18 variants led to missense substitutions altering amino acids highly conserved in orthologues and/or paralogues (Supplementary Fig. 1). Five amino acid substitutions (Met153Ile, Met243Arg, Met305Leu, Gly391Ser and Gly391Asp) were each observed twice, Met305Leu and Met153Ile resulting from different nucleotides substitutions. Three different base changes altering glycine 391 (Gly391Ser, Gly391Cys and Gly391Asp) were observed in five patients.
Clinical and genetic summary of the 19 sporadic patients with de novo HCN1 variants
| Patient number | Gender | Age at the study | Seizure onset | Seizure type at onset | Seizure type at follow-up | Sensitivity to fever | Seizure frequency | Resistance to AEDs | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 12 y 6 m | 12 m | TCS | TCS, At | No | Daily | Yes | Severe ID | EIEE | p.Phe143Tyr |
| 2 | F | 7 y 3 m | 5 m | FS | Fc, TCS | Yes | Weekly to daily | Yes | Mild ID | EIEE | p.Met153Ile |
| 3 | M | 2 y 8 m | 9 m | TCS | TCS, Fc | No | Weekly | Yes | Mild language delay | Uncl epilepsy infantile onset | p.Met153Ile |
| 4 | F | 42 y | 7 m | FS | FS, afebrile TC, Tn and absence | Yes | Last seizure at 20 y | No | Mild ID + autistic traits | GGE-TCS | p.Thr172Pro |
| 5 | F | 17 y | 13 m | FS | FS, afebrile TCS, absence | Yes | R | No | Moderate ID | FS+ | p.Met243Arg |
| 6 | F | 2 y 6 m | 10 m | FS (Tn) | FS | Yes | With fever | No | Normal | FS | p.Met243Arg |
| 7 | F | 2 y | 8 m | FS (Cl) | Cl Gen | Yes | Monthly | No | Normal | FS+ | p.Thr260Ile |
| 8 | M | 5 y 10 m | 9 m | Fc (Cl) | TCS | No | R | No | Normal, mild language delay | Uncl epilepsy infantile onset | p. Ser264Cys |
| 9 | F | 9 y | 42 m | Fc | Fc > TCS; My | No | Daily | Yes | Moderate ID | Childhood focal epilepsy | p.Ile275Thr |
| 10 | F | 12 m | 3 m | TCS | TCS | Yes | 4 m seizure-free | Yes | Mild DD | Uncl epilepsy infantile onset | p.Met305Leu |
| 11 | F | 14 y | 2 m | TCS | TCS, Tn, Fc, Cl | Yes | Monthly | Yes | Severe ID, microcephaly | EIEE | p.Met305Leu |
| 12 | M | Died at 14 m | 30 h | Tn asymmetric, prolonged | Tn asymmetric ± Cl, prolonged apnoea and cyanosis | No | Daily | Yes | Severe ID, microcephaly | NOEE (MMPSI) | p.Gly391Asp |
| 13 | M | Died at 15 m | 48 h | Tn asymmetric, prolonged | Similar but with cyanosis | No | Daily | Yes | Severe ID | NOEE | p.Gly391Asp |
| 14 | F | 6 y 3 m | 5 m | Prolonged FS (20 min) | FS; afebrile TCS: Fc (LOC, hypotonia, cyanosis, vomit) | Yes | Yearly | No | Normal | FS+ | p.Gly391Ser |
| 15 | M | 2 y 5 m | 7 m | HemiCl | Fc, TCS | Yes | Yearly | No | Mild ID | GEFS+ | p.Gly391Ser |
| 16 | F | 29 y | Infancy | Eyelid My | TCS, Fc, absence, My | NA | Monthly to weekly | Yes | Moderate ID, autistic traits | Gen epilepsy with eyelid My | p.Gly391Cys |
| 17 | M | 15 y | 5 m | Hypotonia and cyanosis few h after vaccination | MA; Fc (hypomotor with psychomotor arrest) TCS | Yes | Daily | Yes | Severe ID, autistic traits | EIEE | p.Ile397Leu |
| 18 | M | 7 y | 4 m | Prolonged FS | TCS, with apnoea | Yes | Daily | Yes | Severe ID | EIEE | p.Ser399Pro |
| 19 | M | 15 y | 72 m | Absence | absence + single TCS | No | Daily | Yes | Normal | CAE | p.Arg590Gln |
| Patient number | Gender | Age at the study | Seizure onset | Seizure type at onset | Seizure type at follow-up | Sensitivity to fever | Seizure frequency | Resistance to AEDs | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 12 y 6 m | 12 m | TCS | TCS, At | No | Daily | Yes | Severe ID | EIEE | p.Phe143Tyr |
| 2 | F | 7 y 3 m | 5 m | FS | Fc, TCS | Yes | Weekly to daily | Yes | Mild ID | EIEE | p.Met153Ile |
| 3 | M | 2 y 8 m | 9 m | TCS | TCS, Fc | No | Weekly | Yes | Mild language delay | Uncl epilepsy infantile onset | p.Met153Ile |
| 4 | F | 42 y | 7 m | FS | FS, afebrile TC, Tn and absence | Yes | Last seizure at 20 y | No | Mild ID + autistic traits | GGE-TCS | p.Thr172Pro |
| 5 | F | 17 y | 13 m | FS | FS, afebrile TCS, absence | Yes | R | No | Moderate ID | FS+ | p.Met243Arg |
| 6 | F | 2 y 6 m | 10 m | FS (Tn) | FS | Yes | With fever | No | Normal | FS | p.Met243Arg |
| 7 | F | 2 y | 8 m | FS (Cl) | Cl Gen | Yes | Monthly | No | Normal | FS+ | p.Thr260Ile |
| 8 | M | 5 y 10 m | 9 m | Fc (Cl) | TCS | No | R | No | Normal, mild language delay | Uncl epilepsy infantile onset | p. Ser264Cys |
| 9 | F | 9 y | 42 m | Fc | Fc > TCS; My | No | Daily | Yes | Moderate ID | Childhood focal epilepsy | p.Ile275Thr |
| 10 | F | 12 m | 3 m | TCS | TCS | Yes | 4 m seizure-free | Yes | Mild DD | Uncl epilepsy infantile onset | p.Met305Leu |
| 11 | F | 14 y | 2 m | TCS | TCS, Tn, Fc, Cl | Yes | Monthly | Yes | Severe ID, microcephaly | EIEE | p.Met305Leu |
| 12 | M | Died at 14 m | 30 h | Tn asymmetric, prolonged | Tn asymmetric ± Cl, prolonged apnoea and cyanosis | No | Daily | Yes | Severe ID, microcephaly | NOEE (MMPSI) | p.Gly391Asp |
| 13 | M | Died at 15 m | 48 h | Tn asymmetric, prolonged | Similar but with cyanosis | No | Daily | Yes | Severe ID | NOEE | p.Gly391Asp |
| 14 | F | 6 y 3 m | 5 m | Prolonged FS (20 min) | FS; afebrile TCS: Fc (LOC, hypotonia, cyanosis, vomit) | Yes | Yearly | No | Normal | FS+ | p.Gly391Ser |
| 15 | M | 2 y 5 m | 7 m | HemiCl | Fc, TCS | Yes | Yearly | No | Mild ID | GEFS+ | p.Gly391Ser |
| 16 | F | 29 y | Infancy | Eyelid My | TCS, Fc, absence, My | NA | Monthly to weekly | Yes | Moderate ID, autistic traits | Gen epilepsy with eyelid My | p.Gly391Cys |
| 17 | M | 15 y | 5 m | Hypotonia and cyanosis few h after vaccination | MA; Fc (hypomotor with psychomotor arrest) TCS | Yes | Daily | Yes | Severe ID, autistic traits | EIEE | p.Ile397Leu |
| 18 | M | 7 y | 4 m | Prolonged FS | TCS, with apnoea | Yes | Daily | Yes | Severe ID | EIEE | p.Ser399Pro |
| 19 | M | 15 y | 72 m | Absence | absence + single TCS | No | Daily | Yes | Normal | CAE | p.Arg590Gln |
At = atonic; CAE = childhood absence epilepsy; Cl = clonic; DD = developmental delay; F = female; Fc = focal; FS = febrile seizures; FS+ febrile seizure plus; Gen = generalized; ID = intellectual disability; LOC = loss of consciousness; M = male; MA = myoclonic-atonic; My = myoclonic; NA = unavailable; NOEE = neonatal-onset epileptic encephalopathy; Tn = tonic; TCS = tonic-clonic seizure(s); Uncl = unclassified. Detailed clinical information, including Brain MRI, EEG findings and antiepileptic treatment is provided for Patients 1–19 in Supplementary Table 2.
Clinical and genetic summary of the 19 sporadic patients with de novo HCN1 variants
| Patient number | Gender | Age at the study | Seizure onset | Seizure type at onset | Seizure type at follow-up | Sensitivity to fever | Seizure frequency | Resistance to AEDs | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 12 y 6 m | 12 m | TCS | TCS, At | No | Daily | Yes | Severe ID | EIEE | p.Phe143Tyr |
| 2 | F | 7 y 3 m | 5 m | FS | Fc, TCS | Yes | Weekly to daily | Yes | Mild ID | EIEE | p.Met153Ile |
| 3 | M | 2 y 8 m | 9 m | TCS | TCS, Fc | No | Weekly | Yes | Mild language delay | Uncl epilepsy infantile onset | p.Met153Ile |
| 4 | F | 42 y | 7 m | FS | FS, afebrile TC, Tn and absence | Yes | Last seizure at 20 y | No | Mild ID + autistic traits | GGE-TCS | p.Thr172Pro |
| 5 | F | 17 y | 13 m | FS | FS, afebrile TCS, absence | Yes | R | No | Moderate ID | FS+ | p.Met243Arg |
| 6 | F | 2 y 6 m | 10 m | FS (Tn) | FS | Yes | With fever | No | Normal | FS | p.Met243Arg |
| 7 | F | 2 y | 8 m | FS (Cl) | Cl Gen | Yes | Monthly | No | Normal | FS+ | p.Thr260Ile |
| 8 | M | 5 y 10 m | 9 m | Fc (Cl) | TCS | No | R | No | Normal, mild language delay | Uncl epilepsy infantile onset | p. Ser264Cys |
| 9 | F | 9 y | 42 m | Fc | Fc > TCS; My | No | Daily | Yes | Moderate ID | Childhood focal epilepsy | p.Ile275Thr |
| 10 | F | 12 m | 3 m | TCS | TCS | Yes | 4 m seizure-free | Yes | Mild DD | Uncl epilepsy infantile onset | p.Met305Leu |
| 11 | F | 14 y | 2 m | TCS | TCS, Tn, Fc, Cl | Yes | Monthly | Yes | Severe ID, microcephaly | EIEE | p.Met305Leu |
| 12 | M | Died at 14 m | 30 h | Tn asymmetric, prolonged | Tn asymmetric ± Cl, prolonged apnoea and cyanosis | No | Daily | Yes | Severe ID, microcephaly | NOEE (MMPSI) | p.Gly391Asp |
| 13 | M | Died at 15 m | 48 h | Tn asymmetric, prolonged | Similar but with cyanosis | No | Daily | Yes | Severe ID | NOEE | p.Gly391Asp |
| 14 | F | 6 y 3 m | 5 m | Prolonged FS (20 min) | FS; afebrile TCS: Fc (LOC, hypotonia, cyanosis, vomit) | Yes | Yearly | No | Normal | FS+ | p.Gly391Ser |
| 15 | M | 2 y 5 m | 7 m | HemiCl | Fc, TCS | Yes | Yearly | No | Mild ID | GEFS+ | p.Gly391Ser |
| 16 | F | 29 y | Infancy | Eyelid My | TCS, Fc, absence, My | NA | Monthly to weekly | Yes | Moderate ID, autistic traits | Gen epilepsy with eyelid My | p.Gly391Cys |
| 17 | M | 15 y | 5 m | Hypotonia and cyanosis few h after vaccination | MA; Fc (hypomotor with psychomotor arrest) TCS | Yes | Daily | Yes | Severe ID, autistic traits | EIEE | p.Ile397Leu |
| 18 | M | 7 y | 4 m | Prolonged FS | TCS, with apnoea | Yes | Daily | Yes | Severe ID | EIEE | p.Ser399Pro |
| 19 | M | 15 y | 72 m | Absence | absence + single TCS | No | Daily | Yes | Normal | CAE | p.Arg590Gln |
| Patient number | Gender | Age at the study | Seizure onset | Seizure type at onset | Seizure type at follow-up | Sensitivity to fever | Seizure frequency | Resistance to AEDs | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 12 y 6 m | 12 m | TCS | TCS, At | No | Daily | Yes | Severe ID | EIEE | p.Phe143Tyr |
| 2 | F | 7 y 3 m | 5 m | FS | Fc, TCS | Yes | Weekly to daily | Yes | Mild ID | EIEE | p.Met153Ile |
| 3 | M | 2 y 8 m | 9 m | TCS | TCS, Fc | No | Weekly | Yes | Mild language delay | Uncl epilepsy infantile onset | p.Met153Ile |
| 4 | F | 42 y | 7 m | FS | FS, afebrile TC, Tn and absence | Yes | Last seizure at 20 y | No | Mild ID + autistic traits | GGE-TCS | p.Thr172Pro |
| 5 | F | 17 y | 13 m | FS | FS, afebrile TCS, absence | Yes | R | No | Moderate ID | FS+ | p.Met243Arg |
| 6 | F | 2 y 6 m | 10 m | FS (Tn) | FS | Yes | With fever | No | Normal | FS | p.Met243Arg |
| 7 | F | 2 y | 8 m | FS (Cl) | Cl Gen | Yes | Monthly | No | Normal | FS+ | p.Thr260Ile |
| 8 | M | 5 y 10 m | 9 m | Fc (Cl) | TCS | No | R | No | Normal, mild language delay | Uncl epilepsy infantile onset | p. Ser264Cys |
| 9 | F | 9 y | 42 m | Fc | Fc > TCS; My | No | Daily | Yes | Moderate ID | Childhood focal epilepsy | p.Ile275Thr |
| 10 | F | 12 m | 3 m | TCS | TCS | Yes | 4 m seizure-free | Yes | Mild DD | Uncl epilepsy infantile onset | p.Met305Leu |
| 11 | F | 14 y | 2 m | TCS | TCS, Tn, Fc, Cl | Yes | Monthly | Yes | Severe ID, microcephaly | EIEE | p.Met305Leu |
| 12 | M | Died at 14 m | 30 h | Tn asymmetric, prolonged | Tn asymmetric ± Cl, prolonged apnoea and cyanosis | No | Daily | Yes | Severe ID, microcephaly | NOEE (MMPSI) | p.Gly391Asp |
| 13 | M | Died at 15 m | 48 h | Tn asymmetric, prolonged | Similar but with cyanosis | No | Daily | Yes | Severe ID | NOEE | p.Gly391Asp |
| 14 | F | 6 y 3 m | 5 m | Prolonged FS (20 min) | FS; afebrile TCS: Fc (LOC, hypotonia, cyanosis, vomit) | Yes | Yearly | No | Normal | FS+ | p.Gly391Ser |
| 15 | M | 2 y 5 m | 7 m | HemiCl | Fc, TCS | Yes | Yearly | No | Mild ID | GEFS+ | p.Gly391Ser |
| 16 | F | 29 y | Infancy | Eyelid My | TCS, Fc, absence, My | NA | Monthly to weekly | Yes | Moderate ID, autistic traits | Gen epilepsy with eyelid My | p.Gly391Cys |
| 17 | M | 15 y | 5 m | Hypotonia and cyanosis few h after vaccination | MA; Fc (hypomotor with psychomotor arrest) TCS | Yes | Daily | Yes | Severe ID, autistic traits | EIEE | p.Ile397Leu |
| 18 | M | 7 y | 4 m | Prolonged FS | TCS, with apnoea | Yes | Daily | Yes | Severe ID | EIEE | p.Ser399Pro |
| 19 | M | 15 y | 72 m | Absence | absence + single TCS | No | Daily | Yes | Normal | CAE | p.Arg590Gln |
At = atonic; CAE = childhood absence epilepsy; Cl = clonic; DD = developmental delay; F = female; Fc = focal; FS = febrile seizures; FS+ febrile seizure plus; Gen = generalized; ID = intellectual disability; LOC = loss of consciousness; M = male; MA = myoclonic-atonic; My = myoclonic; NA = unavailable; NOEE = neonatal-onset epileptic encephalopathy; Tn = tonic; TCS = tonic-clonic seizure(s); Uncl = unclassified. Detailed clinical information, including Brain MRI, EEG findings and antiepileptic treatment is provided for Patients 1–19 in Supplementary Table 2.
Clinical and genetic summary of affected members of families with HCN1 variants
| Family/ patient number | Gender | Age at study | Seizure onsset | Seizure type | Seizure at follow-up | AEDs | EEG | Brain MRI | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family T | |||||||||||
| 20 | M | 66 y | 8 m | FS and afebrile TCS | Seizure until age 6 y then single TCS in adult age | PB, currently no AEDs | Normal | NA | NA | GEFS+ | p.Thr171Arg |
| 21 | M | 32 y | 8 m | FS; TCS, possible focal onset >TCS | Seizure-free from age 12 y | PB + GVG + VPA currently no AEDs | Normal | Normal | Borderline | GEFS+ | |
| 22 | F | 21 y | 10 m | FS | FS and afebrile TCS, seizure-free for >3 y | VPA, ESM, currently no AEDs | GSW | Normal | Mild ID (TIQ = 50) | GEFS+ | |
| 23 | F | 21 y | 11 m | FS | FS and afebrile TCS, seizure-free for from 10 to 18 y | VPA | Normal | Normal | Mild ID (TIQ = 66) | GEFS+ | |
| Family M1 | |||||||||||
| 24 | F | 49 y | 3 m | FS, febrile, afebrile TCS | Seizure-free from age 12 y after CBZ | CBZ | NA | NA | Normal | GEFS+ | p.Cys329Ser |
| 25 | F | 29 y | 1 y | FS | Seizure-free after VPA | VPA | NA | NA | Normal | FS | |
| 26 | F | 6 y | 18 m | TCS without fever | Status epilepticus episode treated by PHT then seizure-free on VPA | VPA | Normal | Normal | Normal | GGE | |
| 27 | M | 23 y | 8 m | Febrile TCS | FS and afebrile seizure and absence from age 4 y; non-progressive action myoclonus from age 8 y; seizure-free after VPA | VPA | Normal | Normal | Mild ID (TIQ = 52; VIQ = 46; PIQ = 70) | GGE | |
| 28 | F | 20 y | 13 m | FS | Seizure-free with CBZ until 13 y; CBZ was suspended: seizure recurrence. Seizure-free after CBZ reintroduction | CBZ | Normal | Normal CT scan | Normal | GEFS+ | |
| Family M2 | |||||||||||
| 29 | F | 8 y | 18 m | FS, febrile and afebrile TCS | Seizure-free after VPA | VPA | Fast activity | Normal | Borderline (WIPPSI: IQ = 77) | GEFS+ | p.Val414Met |
| 30 | F | 5 y | 18 m | FS | Seizure-free | No AEDs | Rare focal PA, fast activity | Normal | Borderline (DQ Griffith scale = 70) | GEFS+ | |
| 31 | M | 59 y | 12 m | FS | Seizure-free | No AEDs | NA | NA | Normal | FS | |
| Family O | |||||||||||
| 32 | F | 7 y | 7 y | TCS | Seizure-free | VPA | GSW during IPS | Normal | Normal | GGE | p.Ser680Tyr |
| 33 | F | Unknown | Childhood | Absence | Absence and single TCS in childhood, then seizure-free | No AEDs | GSW | Normal | Normal | CAE |
| Family/ patient number | Gender | Age at study | Seizure onsset | Seizure type | Seizure at follow-up | AEDs | EEG | Brain MRI | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family T | |||||||||||
| 20 | M | 66 y | 8 m | FS and afebrile TCS | Seizure until age 6 y then single TCS in adult age | PB, currently no AEDs | Normal | NA | NA | GEFS+ | p.Thr171Arg |
| 21 | M | 32 y | 8 m | FS; TCS, possible focal onset >TCS | Seizure-free from age 12 y | PB + GVG + VPA currently no AEDs | Normal | Normal | Borderline | GEFS+ | |
| 22 | F | 21 y | 10 m | FS | FS and afebrile TCS, seizure-free for >3 y | VPA, ESM, currently no AEDs | GSW | Normal | Mild ID (TIQ = 50) | GEFS+ | |
| 23 | F | 21 y | 11 m | FS | FS and afebrile TCS, seizure-free for from 10 to 18 y | VPA | Normal | Normal | Mild ID (TIQ = 66) | GEFS+ | |
| Family M1 | |||||||||||
| 24 | F | 49 y | 3 m | FS, febrile, afebrile TCS | Seizure-free from age 12 y after CBZ | CBZ | NA | NA | Normal | GEFS+ | p.Cys329Ser |
| 25 | F | 29 y | 1 y | FS | Seizure-free after VPA | VPA | NA | NA | Normal | FS | |
| 26 | F | 6 y | 18 m | TCS without fever | Status epilepticus episode treated by PHT then seizure-free on VPA | VPA | Normal | Normal | Normal | GGE | |
| 27 | M | 23 y | 8 m | Febrile TCS | FS and afebrile seizure and absence from age 4 y; non-progressive action myoclonus from age 8 y; seizure-free after VPA | VPA | Normal | Normal | Mild ID (TIQ = 52; VIQ = 46; PIQ = 70) | GGE | |
| 28 | F | 20 y | 13 m | FS | Seizure-free with CBZ until 13 y; CBZ was suspended: seizure recurrence. Seizure-free after CBZ reintroduction | CBZ | Normal | Normal CT scan | Normal | GEFS+ | |
| Family M2 | |||||||||||
| 29 | F | 8 y | 18 m | FS, febrile and afebrile TCS | Seizure-free after VPA | VPA | Fast activity | Normal | Borderline (WIPPSI: IQ = 77) | GEFS+ | p.Val414Met |
| 30 | F | 5 y | 18 m | FS | Seizure-free | No AEDs | Rare focal PA, fast activity | Normal | Borderline (DQ Griffith scale = 70) | GEFS+ | |
| 31 | M | 59 y | 12 m | FS | Seizure-free | No AEDs | NA | NA | Normal | FS | |
| Family O | |||||||||||
| 32 | F | 7 y | 7 y | TCS | Seizure-free | VPA | GSW during IPS | Normal | Normal | GGE | p.Ser680Tyr |
| 33 | F | Unknown | Childhood | Absence | Absence and single TCS in childhood, then seizure-free | No AEDs | GSW | Normal | Normal | CAE |
AED = anti-epileptic drug; CBZ = carbamazepine; DQ = developmental quotient; ESM = ethosuximide; F = female; FS = febrile seizure; FS+ = febrile seizure plus; GSW = generalized spike and wave; GVG = vigabatrin; ID = intellectual disability; IPS = intermittent photic stimulation; IQ = intellectual quotient; M = male; NA = unavailable; PA = paroxysmal activity; PB = phenobarbital; PHT = phenytoin; PIQ = performance intellectual quotient; TCS = tonic-clonic seizure(s); TIQ = full-scale intellectual quotient; WIPPSI = Wechsler preschool and primary scale of intelligence; VIQ = verbal intellectual quotient; VPA = sodium valproate.
Clinical and genetic summary of affected members of families with HCN1 variants
| Family/ patient number | Gender | Age at study | Seizure onsset | Seizure type | Seizure at follow-up | AEDs | EEG | Brain MRI | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family T | |||||||||||
| 20 | M | 66 y | 8 m | FS and afebrile TCS | Seizure until age 6 y then single TCS in adult age | PB, currently no AEDs | Normal | NA | NA | GEFS+ | p.Thr171Arg |
| 21 | M | 32 y | 8 m | FS; TCS, possible focal onset >TCS | Seizure-free from age 12 y | PB + GVG + VPA currently no AEDs | Normal | Normal | Borderline | GEFS+ | |
| 22 | F | 21 y | 10 m | FS | FS and afebrile TCS, seizure-free for >3 y | VPA, ESM, currently no AEDs | GSW | Normal | Mild ID (TIQ = 50) | GEFS+ | |
| 23 | F | 21 y | 11 m | FS | FS and afebrile TCS, seizure-free for from 10 to 18 y | VPA | Normal | Normal | Mild ID (TIQ = 66) | GEFS+ | |
| Family M1 | |||||||||||
| 24 | F | 49 y | 3 m | FS, febrile, afebrile TCS | Seizure-free from age 12 y after CBZ | CBZ | NA | NA | Normal | GEFS+ | p.Cys329Ser |
| 25 | F | 29 y | 1 y | FS | Seizure-free after VPA | VPA | NA | NA | Normal | FS | |
| 26 | F | 6 y | 18 m | TCS without fever | Status epilepticus episode treated by PHT then seizure-free on VPA | VPA | Normal | Normal | Normal | GGE | |
| 27 | M | 23 y | 8 m | Febrile TCS | FS and afebrile seizure and absence from age 4 y; non-progressive action myoclonus from age 8 y; seizure-free after VPA | VPA | Normal | Normal | Mild ID (TIQ = 52; VIQ = 46; PIQ = 70) | GGE | |
| 28 | F | 20 y | 13 m | FS | Seizure-free with CBZ until 13 y; CBZ was suspended: seizure recurrence. Seizure-free after CBZ reintroduction | CBZ | Normal | Normal CT scan | Normal | GEFS+ | |
| Family M2 | |||||||||||
| 29 | F | 8 y | 18 m | FS, febrile and afebrile TCS | Seizure-free after VPA | VPA | Fast activity | Normal | Borderline (WIPPSI: IQ = 77) | GEFS+ | p.Val414Met |
| 30 | F | 5 y | 18 m | FS | Seizure-free | No AEDs | Rare focal PA, fast activity | Normal | Borderline (DQ Griffith scale = 70) | GEFS+ | |
| 31 | M | 59 y | 12 m | FS | Seizure-free | No AEDs | NA | NA | Normal | FS | |
| Family O | |||||||||||
| 32 | F | 7 y | 7 y | TCS | Seizure-free | VPA | GSW during IPS | Normal | Normal | GGE | p.Ser680Tyr |
| 33 | F | Unknown | Childhood | Absence | Absence and single TCS in childhood, then seizure-free | No AEDs | GSW | Normal | Normal | CAE |
| Family/ patient number | Gender | Age at study | Seizure onsset | Seizure type | Seizure at follow-up | AEDs | EEG | Brain MRI | Development | Epilepsy type/ syndromes | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family T | |||||||||||
| 20 | M | 66 y | 8 m | FS and afebrile TCS | Seizure until age 6 y then single TCS in adult age | PB, currently no AEDs | Normal | NA | NA | GEFS+ | p.Thr171Arg |
| 21 | M | 32 y | 8 m | FS; TCS, possible focal onset >TCS | Seizure-free from age 12 y | PB + GVG + VPA currently no AEDs | Normal | Normal | Borderline | GEFS+ | |
| 22 | F | 21 y | 10 m | FS | FS and afebrile TCS, seizure-free for >3 y | VPA, ESM, currently no AEDs | GSW | Normal | Mild ID (TIQ = 50) | GEFS+ | |
| 23 | F | 21 y | 11 m | FS | FS and afebrile TCS, seizure-free for from 10 to 18 y | VPA | Normal | Normal | Mild ID (TIQ = 66) | GEFS+ | |
| Family M1 | |||||||||||
| 24 | F | 49 y | 3 m | FS, febrile, afebrile TCS | Seizure-free from age 12 y after CBZ | CBZ | NA | NA | Normal | GEFS+ | p.Cys329Ser |
| 25 | F | 29 y | 1 y | FS | Seizure-free after VPA | VPA | NA | NA | Normal | FS | |
| 26 | F | 6 y | 18 m | TCS without fever | Status epilepticus episode treated by PHT then seizure-free on VPA | VPA | Normal | Normal | Normal | GGE | |
| 27 | M | 23 y | 8 m | Febrile TCS | FS and afebrile seizure and absence from age 4 y; non-progressive action myoclonus from age 8 y; seizure-free after VPA | VPA | Normal | Normal | Mild ID (TIQ = 52; VIQ = 46; PIQ = 70) | GGE | |
| 28 | F | 20 y | 13 m | FS | Seizure-free with CBZ until 13 y; CBZ was suspended: seizure recurrence. Seizure-free after CBZ reintroduction | CBZ | Normal | Normal CT scan | Normal | GEFS+ | |
| Family M2 | |||||||||||
| 29 | F | 8 y | 18 m | FS, febrile and afebrile TCS | Seizure-free after VPA | VPA | Fast activity | Normal | Borderline (WIPPSI: IQ = 77) | GEFS+ | p.Val414Met |
| 30 | F | 5 y | 18 m | FS | Seizure-free | No AEDs | Rare focal PA, fast activity | Normal | Borderline (DQ Griffith scale = 70) | GEFS+ | |
| 31 | M | 59 y | 12 m | FS | Seizure-free | No AEDs | NA | NA | Normal | FS | |
| Family O | |||||||||||
| 32 | F | 7 y | 7 y | TCS | Seizure-free | VPA | GSW during IPS | Normal | Normal | GGE | p.Ser680Tyr |
| 33 | F | Unknown | Childhood | Absence | Absence and single TCS in childhood, then seizure-free | No AEDs | GSW | Normal | Normal | CAE |
AED = anti-epileptic drug; CBZ = carbamazepine; DQ = developmental quotient; ESM = ethosuximide; F = female; FS = febrile seizure; FS+ = febrile seizure plus; GSW = generalized spike and wave; GVG = vigabatrin; ID = intellectual disability; IPS = intermittent photic stimulation; IQ = intellectual quotient; M = male; NA = unavailable; PA = paroxysmal activity; PB = phenobarbital; PHT = phenytoin; PIQ = performance intellectual quotient; TCS = tonic-clonic seizure(s); TIQ = full-scale intellectual quotient; WIPPSI = Wechsler preschool and primary scale of intelligence; VIQ = verbal intellectual quotient; VPA = sodium valproate.
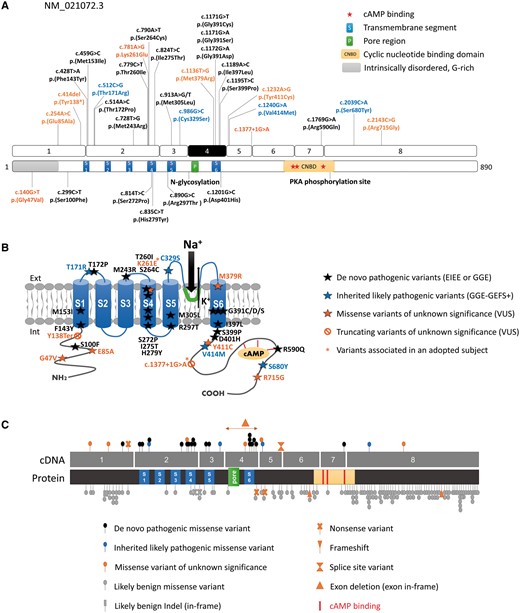
Schematic representation of HCN1 variants on the gene and protein. (A) Location of variants identified in this study (above) or previously reported (Nava et al., 2014; below) on schematic representations of the HCN1 coding exons (NM_021072.3) and corresponding protein domains. Variants in black correspond to de novo variants identified in EIEE or GGE patients. Variants in blue are dominantly inherited within GGE-GEFS+ families. Variants in orange are of unknown significance. (B) Location of HCN1 variants on a schematic representation of the HCN1 channel. Variants indicated with a star are present in an adopted individual with severe EIEE. (C) Comparison of the variants identified in patients with neurodevelopmental phenotypes (above) and variants present in ExAC (below), showing that in control populations, variants clustered in N- and C-terminal regions of the HCN1 protein, which are more tolerant to genetic variation while pathogenic variants tend to cluster to functional or transmembrane domains of the channel.
The spectrum of phenotypic manifestations related to the occurrence of pathogenic and likely pathogenic HCN1 variants is illustrated in Fig. 2A. To display results in a clear and effective way, sporadic and familial cases were analysed separately.
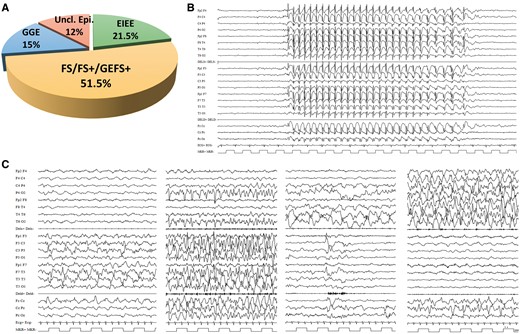
Clinical spectrum associated with HCN1 variants and examples of EEG recordings in HCN1 patients. (A) Graphic representation of the phenotypic spectrum associated with HCN1 variants. (B) EEG recorded in Patient 19 the HCN1 p.Arg590Gln variant, showing typical, generalized 3 Hz spike wave discharges lasting 10 s recorded while the patient was having an absence seizure. (C) Ictal EEG recordings of Patient 12, carrying the p.Gly391Asp mutation, at age 3 months, showing a focal migrating seizure lasting 18 min; seizure starts with a focal paroxysmal ictal discharge (PA) over the left hemisphere (6 min) then the PA migrates to the right hemisphere for additional 12 min. Seizure semiology showed: motor arrest, loss of contact, mild tachycardia, and polypnoea with severe cyanosis and intermittent tonic posturing with vibratory jerking either on the right or the left arm and leg depending upon ictal discharge being to the right or the left hemisphere. From left to right, this panel shows representative fragments of this seizure: PA over the left hemisphere at onset; outbreak of ictal independent yet concomitant PA over the right posterior region (P4-02 and T6-02 leads); shift of the PA from the left to the right; and right-sided PA.
Sporadic epilepsy phenotypes related to de novo pathogenic HCN1 variants
At the time of the study, the 19 probands harbouring de novo HCN1 variants (11 females and eight males) had a median age of 12.0 years (mean age: 13.2, range 1–42 years). Table 1 summarizes the main clinical features of sporadic patients and additional details are provided in Supplementary Table 2. Median age at seizure onset was 7 months (mean age: 11.8 months, range: 30 h–72 months). At onset, seizure types were heterogeneous including mainly generalized episodes, triggered by febrile illness in seven patients (36.8%; Patients 2, 4–7, 14 and 18). Yet, only two of them were reported to have had prolonged febrile seizures and hemiclonic component in one. Six patients (31.5%; Patients 1, 3, 10, 11, 16 and 19) had generalized seizures including absences, eyelid myoclonia and afebrile tonic-clonic seizures. In five probands (26.3%; Patients 8, 9, 12, 13 and 15), initial seizures were classified as possibly focal with a variable combination of symptoms including hypotonia, hypomotor behaviour, apnoea and cyanosis, tonic posturing, clonic jerking with or without secondary generalization. In Patient 17, characterization of the first seizure was difficult and left as unclassified.
At follow-up, all 19 patients had had subsequent polymorphic seizures, either focal or generalized, occurring with variable frequency, from multiple per day (Patients 1, 2, 9, 12, 13 and 16–19) to weekly (Patient 3) to monthly (Patients 7 and 11) or rare (Patients 5, 6, 8, 10, 14 and 15) events. A 42-year-old patient (Patient 4) has been seizure-free and OFF antiepileptic drugs (AEDs) for over 20 years. Patients 12 and 13 died at 14 and 15 months, respectively, due to cardiopulmonary failure, and while living, both had a severe and drug-resistant epilepsy with dozens of daily seizures. Both patients presented worsening of seizure frequency while treated with phenytoin and lacosamide. Overall, at least 10 of 17 living patients (58.8%) with seizures at the time of the study had a drug-resistant epilepsy non-responsive to combinations of several conventional AEDs. The remaining patient with febrile seizures (Patient 6) is currently 2 years and 6 months old and she has seizures only during febrile illnesses.
Following the ILAE guidelines for epilepsy classification (Fisher et al., 2014), three probands (15.7%; Patients 4, 16 and 19) were considered as having GGE including childhood absence epilepsy with typical 3 Hz spike and waves (Fig. 2B), or GGE with tonic-clonic seizures and epilepsy with eyelid myoclonia; five probands (26.3%; Patients 5–7, 14 and 15) were classified within the GEFS+ spectrum; seven probands (36.8%; Patients 1, 2, 11–13, 17 and 18) had a neonatal/infantile onset otherwise unclassified epileptic encephalopathy. The remaining four patients (21%) had infantile onset focal (Patient 9), or infantile onset unclassified epilepsy (Patients 3, 9 and 10).
All patients underwent periodic EEG recordings from seizure onset to current age. According to their epilepsy phenotype, EEG recordings showed either focal, multifocal or generalized paroxysmal activity. Brain MRI was normal or showed non-specific brain abnormalities including parietal, occipital or fronto-temporal atrophy (two patients), thin corpus callosum and mild diffuse white matter hyperintensity (one patient each). Patients 10–12 had acquired microcephaly (Supplementary Fig. 2). Patient 12 underwent several video-EEG monitoring from birth up to age 14 months and numerous focal seizures with an ictal discharge shifting from one hemisphere to the other in the same event were recorded (Fig. 2C). Because of these peculiar migrating seizures, an initial diagnosis of malignant migrating partial seizures of infancy (MMPSI) was suspected, but KCNT1 mutation was ruled out.
Motor and cognitive development were normal in six patients (31.5%; Patients 3, 6–8, 14 and 19) with only mild language developmental delay in two, whereas 13 probands (68.4%) had a variable degree of intellectual disability ranging from mild in four patients (21%; Patients 3, 4, 10 and 15), to moderate in three (15.7%; Patients 5, 9 and 16) and severe intellectual disability in the remaining six (31.5%; Patients 1, 11–13, 17 and 18), with autistic traits in two.
Families with inherited, likely pathogenic HCN1 variants
We identified HCN1 variants segregating with epilepsy in four families (Table 2 and Fig. 3A), for a total of 20 individuals (seven males and 13 females), 14 of whom exhibited an epilepsy phenotype, while six were not known to have had seizures.
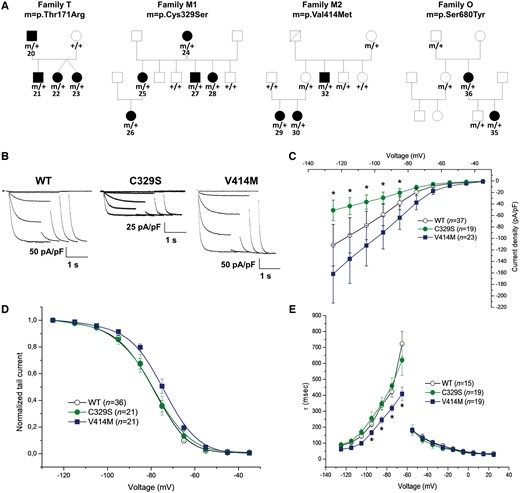
Pedigrees of families with dominantly inherited HCN1 variants and functional impact of variants identified in Families M1 and M2. (A) Pedigrees of Families T, M1, M2 and O. m/+ = the presence of a heterozygous mutation; +/+ = the absence of mutation on both alleles. The patient numbers are indicated for affected individuals and refer to numbering in Table 2. (B) Representative current traces recorded from CHO cells transfected with plasmid expressing human HCN1 wild-type, HCN1 p.Cys329Ser (C329S), or HCN1 p.Val414Met (V414M). From a holding potential of −30 mV, hyperpolarizing steps in the range of −35 to −125 mV followed by a step at −125 mV were applied to measure activation curves in standard two-step protocols. (C) Current density (pA/pF) curve as a function of test voltage for HCN1-wild-type (open circles), C329S (green circles) and V414M (blue squares). (D) Activation curves are presented as normalized tail current obtained for HCN1-wild-type (open circles), C329S (green circles) and V414M (blue squares). (E) Mean activation (left) and deactivation (right), time constant (tau) curves obtained for HCN1-wild-type (open circles), C329S (green circles) and V414M (blue squares). Data are presented as means ± SEM with the numbers of experiments for each condition indicated in parentheses. Statistical analysis was performed by one-way ANOVA with a significance level set to *P < 0.01. Values for half activation potential (V1/2) and inverse slope factor (k) are reported in Supplementary Table 4.
Family T included three siblings and their father, all exhibiting seizures and carrying the Thr171Arg variant. All individuals manifested febrile and afebrile seizures beginning in infancy and childhood with an overall benign outcome. One of the twin girls is still ON valproic acid (VPA) at age 21 years and has yearly seizures; the remaining two siblings and the father have been seizure-free without taking AEDs for several years. The brother and the father had borderline cognitive function, whereas the twin sisters showed mild intellectual disability.
Family M1 included five family members with epilepsy, all carrying the Cys329Ser variant. The proband and four additional symptomatic relatives spread over three generations, exhibited infantile-onset febrile and afebrile tonic-clonic seizures. One individual manifested also a non-progressive action myoclonus; all had normal or borderline cognitive and motor functions. Their EEG recordings and MRI were unremarkable.
Family M2 included six family members carrying the Val414Met variant. The proband, her sister and their maternal uncle presented late infantile, early childhood onset febrile and febrile tonic-clonic seizures. The remaining HCN1 variant carriers were not known to have had seizures. All six family members had normal development and those with seizures had unremarkable MRI and EEG recordings. Only the proband is still on AEDs.
Finally, Family O included five individuals carrying the Ser680Tyr variant although only two were clinically affected. The proband, a female with normal development, presented with a first generalized tonic-clonic seizure at age 7 years and EEG recording showed generalized photoparoxysmal response. Her mother, carrying the same variant had a history of absence seizures in childhood and was treated with valproic acid. The three remaining HCN1 variant carriers were not known to have had seizures.
Thus, the milder outcome and epilepsy phenotypes, including afebrile tonic-clonic seizures and absences with predominant febrile seizures, observed in these four families were consistent with GEFS+ conditions.
Extended HCN1 spectrum: variants of unknown significance
We also collected electroclinical and genetic data of eight probands with variants of unknown significance in HCN1 (Supplementary Table 3 and Supplementary Fig. 3).
A 9-year-old adopted male (Patient 34) with severe EIEE had two predicted damaging variants, one missense (Lys261Glu) in domain S4, and one G>A transition abolishing the donor splice site of intron 5 (c.1377+1G>A). We could not determine whether these variants were located on the same allele (cis) or on different parental alleles (trans) due to absence of DNA from biological parents and unavailability of patient’s material.
Two patients with no history of seizures had de novo HCN1 variants identified by whole exome sequencing or medical exome sequencing. Patient 35 was an 8-year-old male with moderate intellectual disability and acquired microcephaly carrying a de novo nonsense mutation (Tyr138*). Patient 36 had severe intellectual disability and language delay associated with the de novo Met379Arg variant in the S6 transmembrane domain.
Two sporadic female patients had intragenic deletions encompassing exon 4 (encoding the pore of the channel and segment S6), inherited in both cases from their asymptomatic father. Patient 37 had moderate intellectual disability, severe behavioural disturbances (agitation, auto- and hetero-aggressive behaviours) and autism spectrum disorder. Patient 38 had global developmental delay, behavioural disturbances with intolerance to frustration, and peculiar facial features (long face, arched eyebrows, marked but short philtrum, and epicanthus). This latter patient had a HCN1 deletion extending to exon 5 and also carried a de novo 339 kb 17q12 duplication [arr[hg19] 17q12(37332899_37672040)] of unknown significance.
Finally, three sporadic patients (Patients 39–41) with mild GGE and GEFS+ phenotypes, carried HCN1 missense variants inherited from an asymptomatic parent. Proband 39 had the Glu85Ala variant, inherited from his asymptomatic mother, and exhibited GGE with tonic-clonic seizures at age 11 years and generalized spike wave discharges enhanced during intermittent photic stimulation. He also manifested cortical tremor with giant evoked potentials and positive C-reflex. Similarly, a 15-year-old female (Proband 41) carrying the Arg715Gly variant inherited from her asymptomatic father, exhibited absence seizures with 3 Hz spike-wave discharges on EEG recordings at age 7 years leading to a diagnosis of childhood absence epilepsy. The remaining proband had infantile onset of febrile and afebrile tonic-clonic seizures suggestive of a GEFS+ phenotype and carried the Tyr411Cys variant (Proband 40), inherited from his asymptomatic father.
Genotype-phenotype correlations
We investigated the correlations existing between variants and their associated phenotypes further. Twelve of 14 de novo pathogenic missense variants clustered in transmembrane domains of HCN1 including S1, S4, S5 and S6 segments or were located in close proximity to domains S3/S6 (Fig. 1A and B), whereas the four missense variants identified in families were all located outside transmembrane segments, either in extracellular loops (S1-S2: T171R; S5-P: C329S) or in the intracellular C-terminal domain (V414M, S680Y). Of the two de novo variants located outside transmembrane domains, one (R590Q) affected a residue of the cyclic nucleotide-binding domain (CNBD) critical for cAMP binding (Zagotta et al., 2003; Lolicato et al., 2011) and was associated with a mild generalized epilepsy phenotype. All four variants of unknown significance identified in patients with mild phenotypes, although predicted to be damaging, were also located outside transmembrane domains, either in the N or C-terminal regions. Altogether, these observations suggested that variants located in transmembrane segments or domains required for formation of the pore structure are generally associated with more severe phenotypes than variants located in extracellular loops or N/C-terminal domains.
Patients with identical de novo variants strikingly had concordant phenotypes, indicating that the phenotype was largely determined by the mutation itself: Patients 2 and 3 (M153I) both exhibited an infantile onset epilepsy, although Patient 2, who was older, also had seizures increased during febrile illness and mild intellectual disability; Patients 5 and 6 (M243R) had fever-related seizures; Patients 14 and 15 (G391S) both had infantile onset of febrile and afebrile seizures; Patients 12 and 13 with G391D showed neonatal epileptic encephalopathy with severe, intractable seizures and profound cognitive and motor delay leading to death at ages 14 and 15 months, respectively. The concordance of phenotypes in Patients 10 and 11, who had the same M305L variant (as the result of two different base changes) was less obvious, probably because Patient 10 was an infant whereas Patient 11 was an adolescent. Yet, both patients had fever-sensitive seizures and similar postnatal microcephaly (Supplementary Fig. 2).
Functional impact of inherited, likely pathogenic variants
To determine the functional impact of the variant on the biophysical properties of the channel, we performed whole-cell patch-clamp recordings in CHO cells for two of four inherited, likely pathogenic HCN1 variants. HCN1 C329S exhibited a smaller current density compared with the wild-type channel (Fig. 3B and C), but comparable voltage-dependence of activation (Fig. 3D). The activation and deactivation kinetics were not altered (Fig. 3E). Conversely, HCN1 V414M showed a current density similar to the wild-type channel (Fig. 3B and C) but the half-activation voltage (V1/2) was shifted to the right by ~5 mV with faster activation kinetics at all tested voltages (Fig. 3D and E). These results suggest that these two familial variants behave as mild loss- or gain-of-function of the homotetrameric HCN1 channels, respectively.
Functional impact of de novo pathogenic variants
We then carried out whole-cell patch-clamp recordings in CHO cells transiently transfected with wild-type HCN1 or mutant HCN1 channels corresponding to 10 selected de novo HCN1 variants. No Ih current was recorded from cells transfected with M305L, G391D and S399P and rarely from cells transfected with K261E (Fig. 4A–C), indicating a probable loss-of-function for these mutants. Recordings were inconstant in CHO cells for G391C, G391S and I397L, preventing a detailed analysis (Fig. 4E). All mutant channels leading to a loss-of-function were expressed and reached the plasma membrane although their quantity was highly decreased for all except G391C, with the lowest amounts observed for K261E and M305L (Supplementary Fig. 4). On the contrary, voltage-dependent, slowly activating inward currents were recorded upon hyperpolarization in cells transfected with wild-type, M153I, M243R and R590Q channels, consistent with an expression of functional channels (Fig. 4A and B). Current densities were significantly decreased for M243R and R590Q compared to the wild-type channel. M153I had major effects on channel gating (Fig. 4D), as the half-activation voltage was shifted to the depolarizing direction by ~36 mV compared to wild-type channel. The M153I variant also resulted in significantly faster activation and slower deactivation kinetics than wild-type HCN1 (Fig. 4E). In contrast, the half-activation voltage of R590Q was shifted to the hyperpolarizing direction by ~9 mV, without significant effect on the activation and deactivation kinetics. To mimic the heterozygous state of the mutation, we performed co-expression of wild-type HCN1 and the more interesting variant, M153I. The shift of the half-activation voltage was intermediate between wild-type and homomeric channels, shifted to the depolarizing direction ~12 mV compared to wild-type channel. These results largely confirmed that both de novo and inherited HCN1 variants associated with epilepsy may indifferently lead to loss- or gain-of-function of homotetrameric HCN1 channel properties in heterologous cells, without any immediately obvious correlation between phenotypes and the effects on homomeric channels.
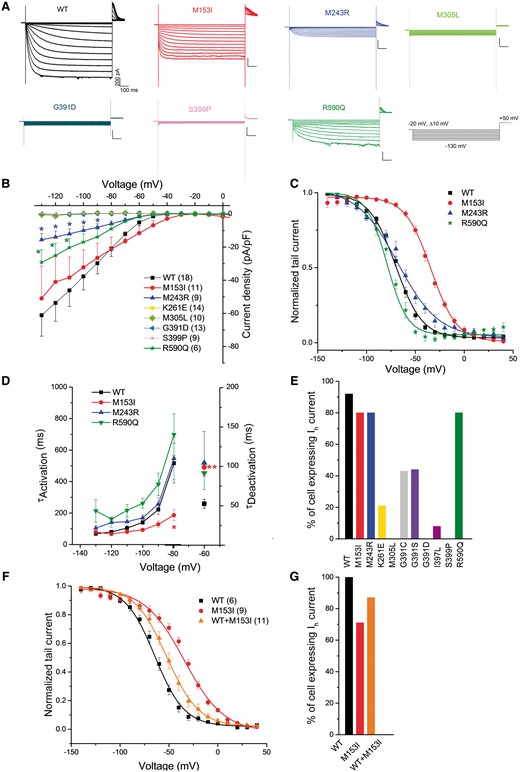
Functional impact of selected de novo HCN1 variants using whole-cell patch-clamp. (A) Representative traces of whole-cell currents recorded in CHO cells transfected with constructs for wild-type (WT), M153I, M243R, M305L, G391D, S399P or R590Q human HCN1 channels. Currents were elicited by test pulses ranging from −20 mV to −130 mV in 10 mV increments from a holding potential of −20 mV. (B) Plot of mean current density as a function of test voltage for wild-type, M153I, M243R, K261E, M305L, G391D, S399P and R590Q human HCN1 channels (two-way ANOVA, *P < 0.05). (C) Mean tail current activation curves for wild-type, M153I, M243R, and R590Q HCN1 channels. (D) M153I mutant channels have faster activation time constants than wild-type HCN1 channel (two-way ANOVA, *P < 0.05). The M153I mutant channels also display significantly increased deactivation time constants compared with those of wild-type HCN1 (one-way ANOVA, **P < 0.01 for voltage −60 mV). (E) Plot of the per cent of cells expressing the Ih current for all variants. (F) Mean tail current activation curves for wild-type, M153I and wild-type/M153I HCN1 channels. (G) Plot of the per cent of cells expressing the Ih current for all variants tested. Data are presented as means ± SEM with the numbers of experiments for each condition indicated in parentheses. Values for half activation potential (V1/2) and inverse slope factor (k) are reported in Supplementary Table 4.
To gain further insight into the mechanisms by which HCN1 variants cause divergent phenotypes, M305L, G391S, G391C G391D and I397L mutant channels were expressed alone or in combination with the wild-type channel to study the behaviour of homo- and heterotetrameric channels in human HEK293 cells, as we found that these mutants express more robustly in this cell line compared to CHO cells, allowing a more detailed characterization (Fig. 5).
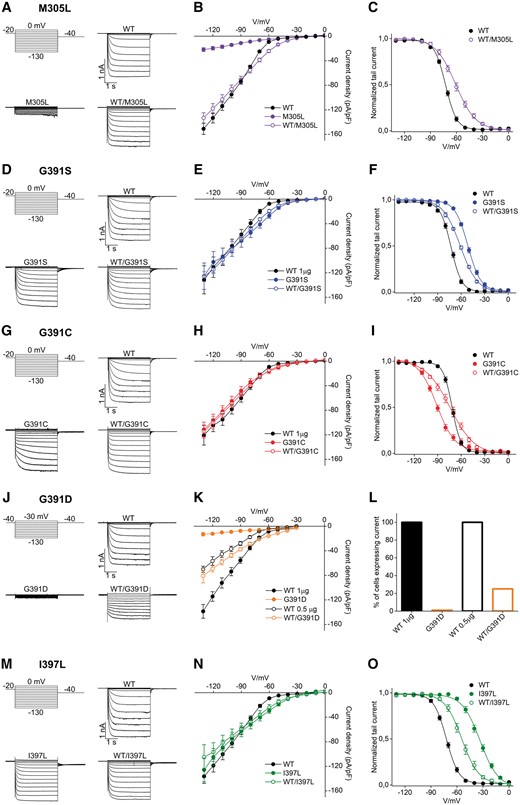
Functional impact of variants altering glycine 391, isoleucine 397 and methionine 305 on HCN1 homotetramers and heterotetramers. Representative current traces recorded by whole-cell patch-clamp using the indicated voltage step protocol from HEK293 cells transiently transfected with 1 µg of plasmid DNA of HCN1 wild-type, M305L (A), G391S (D), G391C (G), G391D (J), and I397L (M) channels, as indicated. Mean steady state current/voltage relationship (I/V) from cells transfected with wild-type channel (filled black circles); wild-type/M305L and M305L (open and filled violet circles) (B), wild-type/G391S and G391S (open and filled blue circles) (E), wild-type/G391C and G391C (open and filled red circles) (H), wild-type/G391D and G391D (open and filled orange circles) (K) or wild-type/I397L and I397L (open and filled green circles) (N). Mean activation curves of wild-type, M305L (C), G391S (F), G391C (I) and I397L (O) channels, both in homo- and heterotetrameric conditions. Lines show data fit to a Boltzmann function yielding half activation potential (V1/2) and inverse slope factor (k) values reported in Supplementary Table 4. (L) Histogram showing the percentage of cells expressing current of wild-type (100%), G391D (0%) and wild-type/G391D (25%). The percentage of expression for all other variants are shown in Supplementary Fig. 5. All values are reported as mean ± SEM.
When the mutant HCN1 subunits were expressed alone, a current was recorded for M305L, G391S, G391C and I397L, but not for G391D (Fig. 5A, D, G, J and M). When the wild-type and mutant forms were co-transfected, all the variants gave rise to measurable currents (Fig. 5A, D, G, J and M). The amount of current recorded from G391S, G391C, I397L either alone or co-transfected with the wild-type, was comparable to the wild-type condition (Fig. 5E, H and N). Interestingly, the analysis of the voltage dependence of G391S and G391C channels (Fig. 5F and I) indicates a strong but opposite effect of the two variants on V1/2. G391S induced a right shift of +21 mV (Fig. 5F), while mutation G391C led to a left shift of −19 mV (Fig. 5I). The behaviour of the heteromeric channels was intermediate in both cases, with respective shifts of +11.5 mV (G391S) and −4 mV (G391C). I397L showed the strongest shift of the V1/2 (+40 mV) and, similarly to G391S and G391C, the co-expression with the wild-type subunit conferred an intermediate phenotype with a shift of the V1/2 of +20 mV (Fig. 5O).
In contrast, the M305L substitution caused a decrease in the number of cells that were expressing current, with 45% of measured cells showing a strongly reduced current density (Fig. 5B), precluding the analysis of tail currents. Interestingly, the co-expression of mutant and wild-type subunits rescued this current loss, returning the current density values and the number of cells expressing current to the range of the wild-type condition (Fig. 5B and Supplementary Fig. 5). However, the analysis of the voltage dependence of this heteromeric channel showed a right shift of the V1/2 of +12 mV and an increase in slope factor (Supplementary Table 4), indicating different gating properties than the wild-type channel (Fig. 5C).
A more severe picture emerged for the G391D mutant. On one hand, both the lack of current in the mutant and the halved current of the co-expression (roughly comparable to half dose of the wild-type, Fig. 5K), suggested a possible lack of expression for this mutant. On the other hand, the G391D/wild-type currents showed features that were clearly specific to the co-expression of wild-type/G391D subunits, such as the presence of an instantaneous component (Fig. 5J and K) and a strong reduction (80%) in the number of transfected cell expressing a measurable current (Fig. 5L), suggesting that the heteromeric channel can form and, although poorly, conduct current.
Impact of G391D on channel structure
To understand the impact of the G391D mutation associated with the most severe neonatal epileptic encephalopathy phenotype on HCN1 protein structure/function, we performed molecular dynamic simulations on the transmembrane domains of wild-type and mutant channels. The G391D variant generated an opening of the inner channel gate by ~1.8 Å during the first 25 ns of simulation. This favoured an influx of water into the channel cavity, which was absent in the wild-type channel (Fig. 6A, Supplementary Fig. 6A and B). The presence of carboxyl groups in mutant channels caused a strong binding of potassium ions to one or multiple aspartate 391 residues (Fig. 6B). While this position was not occupied by a cation in the wild-type channel (Fig. 6E), the mutant remained in this open- but non-conducting conformation for the whole 100 ns of simulation (Fig. 6F). The results of these simulations are consistent with the electrophysiological data, which showed that the homomeric channels are non-conductive, presumably because the ionic path is blocked by cations firmly bound to Asp391. In addition, they correspond to previous findings on HCN global dynamics (Weissgraeber et al., 2017). A similar inhibition of channel conductance by an ion, which is complexed by negative charges at the entry into the cavity, was also reported previously (Tayefeh et al., 2007).
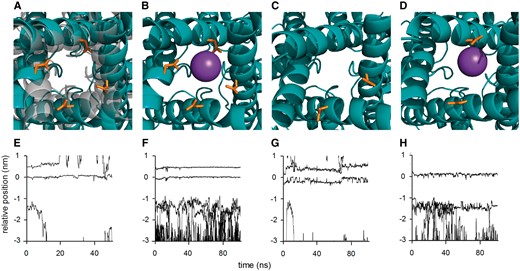
Molecular dynamic simulation of the effect of G391D on HCN1 channel structure. Results of molecular dynamic simulations of HCN1 wild-type and G391D mutant channel in POPC membrane: (A) Overlay of snapshots from S6 domains in bottom view perspective from the cytosol towards channel cavity for wild-type (grey) and homozygous G391D mutant (teal blue) after a total simulation time of 25 ns. (B) Snap shot of homozygous mutant with K+ ion (sphere) complexed by Asp391 (orange). (C) Snap shot of heterozygous mutant with Asp391 in two adjacent monomers without bound K+ ion. (D) Snap shot of heterozygous mutant with Asp391 in two opposite monomers with bound K+ ion. Lower row shows trajectories of potassium ions along the channel axis of HCN1 in wild-type channel (E), homozygous G391D mutant (F), heterozygous mutant with G391D in two adjacent monomers (G), and heterozygous mutant with G391D in two opposite monomers (H). The K+ binding site S4 in the selectivity filter is located at 0 nm while residue 391 is at ~−1.5 nm. For clarity, only a subset of all ions is shown.
We then simulated the behaviour of the heteromeric HCN1 channels in which two of the four subunits carried the G391D variant. The simulation showed that channels in which the mutation was introduced in opposite monomers behave like the homomeric mutants; water enters the cavity and K+ ions associate to Asp391, blocking conductance of the channel (Fig. 6D and H). In contrast, when the mutation was introduced in adjacent subunits, the channel remained closed. There was no entry of water and no blockage by K+ ions during 100 ns of simulation (Fig. 6C and H). These results are also in agreement with the electrophysiological recordings, which showed that some functional HCN1 type currents can be measured in a few cells expressing a mixture of wild-type and mutant channels. It is interesting to note that the helices, which carried the mutation in adjacent monomers showed small deformations (Fig. 6C). It is therefore tempting to speculate that this small widening of the inner gate, which is caused by the greater space requirement of the aspartate side chain, is responsible for the large instantaneous current observed for the heteromeric channels.
Discussion
HCN1 spectrum
Previously, we described five de novo HCN1 missense variants in patients with a Dravet-like phenotype (Nava et al., 2014a), recognized as early infantile epileptic encephalopathy-24 (EIEE24; OMIM #615871). Since then, a single independent study has reported a de novo HCN1 variant (p.Ala387Ser) in a patient with Rett-like syndrome (Lucariello et al., 2016). The clinical spectrum associated with HCN1 variants has thus remained largely unknown. The analysis of the electroclinical features of 33 patients harbouring de novo and inherited heterozygous HCN1 variants uncovered a wide phenotypic spectrum ranging from mild GGE and GEFS+ phenotypes, to infantile onset focal or unclassified epilepsies to catastrophic epilepsies assembled under the term of ‘neonatal/infantile epileptic encephalopathies’. One of two patients with neonatal onset epileptic encephalopathy carrying the G391D variant, exhibited video-EEG recorded prolonged focal seizures with an ictal discharge shifting from one hemisphere to the other within the same event, suggesting a diagnosis of MMPSI. This study also provides further evidence that there is a biological and clinical continuum between mild, benign generalized epilepsies and severe epileptic encephalopathies, as the result from genetic variants in the same gene, as previously observed for other genes encoding ion channels (e.g. SCN1A, SCN2A and KCNQ2). In our cohort, 36.8% of the sporadic and ~20% of the whole cohort exhibited severe epilepsy corresponding to EIEE whereas the great majority of both sporadic (42%) and familial cases manifested a milder phenotype comprehensive of a less severe epilepsy, associated with mild intellectual disability or normal cognition. Indeed, >65% of our patients manifested febrile seizures, febrile seizure plus or GGE including childhood absence epilepsy with normal or borderline cognitive and motor functions.
The electroclinical features of cortical tremor with photosensitivity and of non-progressive action myoclonus in two individuals with HCN1 variants corroborates the hypothesis of neuronal hyperexcitability, possibly causing a depolarized resting membrane potential in neurons that are more prompt to firing. We speculate that this effect in the thalamocortical network similarly results in abnormal generalized spike-wave activity, a hallmark of GGE, observed in patients with HCN1 variants. HCN1 is also moderately expressed in the heart (source: GTEX). Yet, contrary to patients with HCN4 pathogenic variants (DiFrancesco, 2013), no obvious cardiac abnormalities have been yet reported in patients with HCN1 variants. This is an important aspect that should also be accounted for in further studies.
Genotype-phenotype correlations and impact of HCN1 pathogenic variants
Overall, HCN1 variants causing severe phenotypes tended to cluster within or close to transmembrane domains and have stronger impact on channel function whereas variants segregating with milder phenotypes were located in extracellular loops or in the intracellular N- and C-terminal parts of the channel and had milder effects on Ih current. Additionally, noticeable phenotypic concordance was observed in pairs of patients harbouring identical de novo HCN1 variants, indicating that the epilepsy phenotype is largely determined by this single variant. In particular, G391D observed in two unrelated patients, one of Italian origin and the other Portuguese, caused an almost identical catastrophic neonatal phenotype. Both probands presented with severe, intractable epileptic encephalopathy with daily seizures from the first days of life, and never acquired psychomotor skills. Seizure semiology was also similar with asymmetrical tonic posturing and predominant dysautonomic signs including apnoea and cyanosis. Two patients with a different change (G391S) altering the same amino acid both showed milder phenotypes consistent with GEFS+ spectrum. The three different changes altering glycine 391 show that the phenotype is not only determined by the position of the altered amino acid on the channel but also by the nature and consequence of the amino acid change. The impact of G391S/C, associated with milder phenotypes, was limited to shifts in the voltage dependency of the channel, whereas G391D, causing the most severe phenotype, corrupted channel function in a more complex manner: wild-type/mutant heteromeric channels exhibited a strong reduction in current density with a high instantaneous activating current component. Molecular dynamic simulation revealed that the aspartate at the entrance to the inner pore cavity electrostatically interacts with cations preventing, in the homozygous condition, or altering, in the heterozygous state, their permeation. These results show that different amino acid substitutions at a critical position can either reduce (loss-of-function) or increase (gain-of-function) HCN1 homomer conductance. However, mutation spectrum and co-expression studies suggest that seizures are specific to variants leading to shifts in heteromeric channel properties that could also be interpreted as a gain-of-function impact. We hypothesize that variants leading to a gain-of-function lead to neuronal hyperexcitability sustaining seizures, while variants leading to haploinsufficiency of HCN1 result, on the contrary, in neuronal hypoexcitability and predispose to intellectual disability/autism spectrum disorder, as previously demonstrated for SCN2A (Ben-Shalom et al., 2017; Wolff et al., 2017). More suitable neuronal and animal models are needed to fully understand the complexity of the effects resulting from HCN1 dysfunction related to epilepsy and intellectual disability.
Variants of unknown significance
The observation of an adopted child with severe EIEE harbouring both missense (p.Lys261Glu) and splice site (c.1377+1G>A) variants suggested that this severe phenotype could be associated with recessive inheritance. Functional analysis of p.Lys261Glu further confirmed that it leads to a probable loss-of-function of homomeric channels. Yet, the phase of the variants (i.e. location in cis or in trans) could not be determined due to the unavailability of parental and patient’s material. Further evidence is therefore needed to conclude on the existence of recessive HCN1 disorders. The contribution of variants introducing premature termination codon (including the de novo nonsense variant identified in this study) and of intragenic deletions (e.g. deletion of exon 4 described here in two patients), which are likely to lead to heterozygous loss-of-function of the mutated allele remains unclear. The same applies to de novo variants associated with intellectual disability/autism spectrum disorder phenotypes without seizures. For this reason, we classified these variants as variants of unknown significance, although they probably contribute to intellectual disability/autism spectrum disorder in the patients described in this study. Patients with autism spectrum disorder with HCN1 variants could be related to the interaction of HCN1 channels with SHANK3, a scaffolding protein highly enriched in the post-synaptic density of glutamatergic synapses, whose genetic alterations have been linked to autism spectrum disorder (Yi et al., 2016; Zhu et al., 2018). Finally, HCN1 missense variants located in parts of the channel more tolerant to variations (e.g. N-/C-terminal domains) seem associated with a lower penetrance of epilepsy/intellectual disability phenotypes. Inherited HCN1 variants of unknown significance described in this study could even act as susceptibility factors and interact with variants in other genes to determine occurrence of polygenic epilepsy, as described in the WAG/Rij rat (Wemhoner et al., 2015).
In conclusion, this study provides a comprehensive delineation of the phenotypic spectrum associated with HCN1 variants ranging from GEFS+ or GGE to neonatal or infantile epileptic encephalopathy. Recurrent variants showed striking genotype-phenotype correlations. HCN1 pathogenic variants might be either de novo or dominantly inherited and lead to gain-of-function or dominant negative effects underlying neuronal hyperexcitability in various neuron types including the thalamocortical network, thus generating aberrant generalized spike-waves as observed in our patients. Animal models reproducing HCN1 variants should help understanding more accurately the associated pathophysiological mechanisms in the hope to develop more adapted treatments for this group of disorders.
Abbreviations
- EIEE
early infantile epileptic encephalopathy
- GEFS+
genetic generalized epilepsy with febrile seizure plus
- GGE
genetic generalized epilepsy
- HCN
hyperpolarization-activated, cyclic nucleotide-gated
Acknowledgements
We thank the patients and their family for their participation in this study, Dr. Juliane Stieber (Universität Erlangen-Nürnberg, Germany) for providing plasmids containing the human HCN1 cDNA and Prof. Dirk Isbrandt (German Center for Neurodegenerative Diseases (DZNE) and University of Cologne) for critical reading of the manuscript and interesting discussions. Calculations required for MD simulations were conducted on the Lichtenberg high performance computer of the TU Darmstadt. The Mariani Foundation, which promoted and organized the International Workshop on the Genetic Epileptic Encephalopathies, Florence 2016, is also acknowledged. This Workshop was the starting point of an international collaboration leading to the creation of the HCN1 consortium. Data collection and generation within this fruitful international collaboration has led to this manuscript. The research leading to these results was partly generated on the electrophysiology core facility of the institute of brain and spine (ICM, Paris). This study also makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from [email protected]. Funding for the project was provided by the Wellcome Trust.
Funding
The research generating these results was funded by the Agence National de la Recherche (ANR EUHFAUTISM), the ‘Investissements d’avenir’ program ANR-10-IAIHU-06 (IHU-A-ICM), the Bio-Psy labex, INSERM, the Assistance Publique des hôpitaux de Paris (AP-HP), the Pierre and Marie Curie University, the Italian Ministry of Health (Ricerca Finalizzata Giovani Ricercatori), projects GR-2010-2304834 to J.C.D. and A.B. and GR-2016-2363337 to J.C.D., the Ministry of Research and the Arts (HMWK) of the Hessen state (LOEWE project iNAPO), the Fondazione CARIPLO grant 2014-0796 to A.M. and B.S., and the Telethon Exploratory Project GEP14137 to A.M. This work was supported by a grant from the European Commission Seventh Framework Programme FP7 under the project DESIRE (grant agreement no. 602531).
Competing interests
S.I is employed by Ambry Genetics and F.K. is medical director of Mendelics Genomic Analysis, companies that provide gene panel and medical exome sequencing as commercially available tests. All other authors report no competing interests.
Appendix 1
Collaborators (ULB)
Vilain Catheline (Department of Genetics, Hôpital Universitaire des Enfants Reine Fabiola, ULB Center of Human Genetics; Department of Genetics, Hôpital Erasme; and Interuniversity Institute of Bioinformatics in Brussels at the Université Libre de Bruxelles, Brussels, Belgium); Bouysran Youssef (Department of Genetics, Hôpital Erasme and Interuniversity Institute of Bioinformatics in Brussels at the Université Libre de Bruxelles, Brussels, Belgium); Aeby Alec (Department of Pediatric Neurology, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, Brussels, Belgium).
References
Author notes
Carla Marini, Alessandro Porro, Agnès Rastetter and Carine Dalle authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}