




This scientific commentary refers to ‘Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism’, by Puschmann et al. (doi:).
The pathogenesis of Parkinson’s disease, in keeping with other common neurodegenerative diseases, is a complex interplay of genetic and environmental factors. Significant progress has been made in dissecting the genetic architecture of both rare monogenic and more common sporadic forms of the disease. Mutations in three genes, SNCA, LRRK2 and VPS35, cause autosomal dominant Parkinson’s disease, while mutations in six genes cause autosomal recessive Parkinson’s disease/parkinsonism: PINK1, PARK7/DJ-1, PARK2/PARKIN, PLA2G6, ATP13A2, and FBXO7. Taken together, monogenic forms of Parkinson’s disease account for 30% of cases of familial disease and 3–5% of sporadic disease. Nonetheless much of our knowledge of the pathogenic pathways that lead to neuronal dysfunction and death has emerged from the identification of these genes. The discovery of the monogenic forms naturally posed the question of whether these genes also influence the risk of sporadic disease. If the same genes that can harbour highly penetrant mutations in Parkinson’s disease also contain risk variants for sporadic disease, this would provide a genetic and pathophysiological link between the mendelian and sporadic forms (Lesage and Brice, 2012). Several genome-wide association studies have independently identified SNCA and LRRK2 as risk loci for sporadic disease, thus linking the risk of sporadic Parkinson’s disease to that of the autosomal dominant forms (Nalls et al., 2014). However, the mechanism by which a risk variant alters biological function and confers susceptibility to disease remains an essential missing piece of the puzzle. In this issue of Brain, Puschmann and co-workers examine whether a genetic variant in PINK1 contributes to the risk of sporadic disease, and explore the biological mechanism by which it may do so (Puschmann et al., 2016).
Homozygous and compound heterozygous mutations in PINK1 are the second most frequent cause of autosomal recessive Parkinson’s disease. PINK1-associated autosomal recessive Parkinson’s disease resembles sporadic Parkinson’s disease with an akinetic rigid syndrome with good response to levodopa, levodopa-induced dyskinesias, and occasionally dystonia. Many heterozygous putative pathogenic mutations in PINK1 have been observed in sporadic and familial Parkinson’s disease, which has led to the hypothesis that they may be a susceptibility factor in the development of the sporadic form. However, heterozygous PINK1 mutations have also been discovered in apparently healthy controls, and relatives who are heterozygous carriers in pedigrees with PINK1 homozygous mutations do not always show parkinsonism (Klein et al., 2007). If the hypothesis is true, one would expect heterozygous mutations to be more common in patients than in controls; the mean age of onset would be predicted to be intermediate between the state of the homozygous mutations and no mutation; there may be preclinical manifestations of disease in neuroimaging of heterozygous mutation carriers; and there should be evidence that the heterozygous mutation affects the biological function of PINK1, either through haploinsufficiency, or through dominant negative or dominant effects.
This study focuses on one particular PINK1 variant p.G411S, which has occasionally shown dominant inheritance, leading Puschmann et al. to propose that a single risk allele may present sizeable risk and may have a pathological effect distinct from other variants. They performed a case control study on five different Caucasian series, studying a total of 2560 patients with Parkinson’s disease and 2145 controls. Heterozygous PINK1 p.G411S variants were present in 19 cases (0.74%) and five controls (0.23%), confirming a significant association of the heterozygous p.G411S variant with risk of Parkinson’s disease with an odds ratio of 2.92 (P = 0.032), which remained significant in a meta-analysis incorporating previous published studies. Interestingly the median age of disease onset was significantly younger in the PINK1 heterozygous carriers, compared to non-carrier status (59 years versus 64 years, P = 0.012). These findings are in contrast to the largest previous meta-analysis (Marongiu et al., 2008), which studied 1100 patients and 400 controls for heterozygous variants in the PINK1 gene and reported no significant difference in frequencies (1.8% versus 1.5%) between cases and controls. However, this study was smaller than that of Puschmann et al. and considered all PINK1 variants rather than only the p.G411S variant. Variation at the PINK1 locus was not identified in genome-wide association study meta-analyses either, although this may be due to the low frequency of the p.G411S variant in non-Scandinavian populations.
Puschmann et al. then address how the p.G411S variant exerts its effect on PINK1. PINK1 is a ubiquitously expressed serine/threonine kinase with a mitochondrial targeting sequence. Relevant to this study is the emergence of a critical role for PINK1 in the regulation of mitochondrial quality control through the selective elimination of damaged mitochondria via mitophagy (Pickrell and Youle, 2015). Mitochondrial depolarization leads to accumulation of a PINK1 dimer on the outer membrane of damaged mitochondria. Crucially PINK1 phosphorylates residue Ser65 of ubiquitin (p-ser65-ub) and ubiquitin chains conjugated to mitochondrial proteins (Durcan and Fon, 2015), increasing the level of p-ser65-ub from undetectable to 20% of total ubiquitin in mitochondria (Ordureau et al., 2014). One function of p-ser65-ub is to serve as a receptor for binding and recruitment of autophagy receptors and parkin (Lazarou et al., 2015). Together with the PINK1-dependent Ser65 phosphorylation of the Ubl domain in parkin, binding of p-ser65-ub to parkin converts it from the inactive conformation to the fully active conformation. This results in ubiquitination of many outer mitochondrial membrane proteins by the parkin E3 ligase, and the triggering of mitophagy.
Puschmann et al. first analysed skin fibroblasts, and induced neurons, from two patients with heterozygous p.G411S, and compared them to controls, and furthermore to cells with the Q456X homozygous (patients) and Q456X heterozygous mutations (unaffected carriers). The Q456X mutation provides a useful comparison as in the homozygous state the PINK1 protein is absent and thus kinase function is absent, while in the heterozygous state, intermediate levels of PINK1 protein are present; if the heterozygous state is functionally significant, the effect must reflect haploinsufficiency. Of note, levels of PINK1 protein are the same in the heterozygous p.G411S state as in controls. Next, as a measure of the kinase function of PINK1, Puschmann et al. tested phosphorylation of ser65-ubiquitin using two methods: an ELISA-based approach and an automated high content imaging approach. Using prolonged treatment with valinomycin to induce mitochondrial depolarization, and subsequent mitophagy, they demonstrate that the p-ser65-ub signal, and hence PINK1 kinase activity, is significantly reduced in cells with p.G411S heterozygous compared to controls or the Q456X heterozygous state. This confirms that the p.G411S heterozygous state does indeed result in impaired PINK1 function in neurons, but that the effect is not recapitulated in a model of haploinsufficiency.
To dissect the mechanism of action of the p.G411S variant, the authors performed theoretical molecular modelling of the PINK1 protein, and molecular dynamics simulation of the effect of the p.G411S variant. In the PINK1 monomer, p.G411S induces mispositioning of ubiquitin in the active site, which increases the distance between the Ser65 residue on ubiquitin and ATP, and reduces kinase activity. However, the modelling of the heterodimer formed between wild-type PINK1 and PINK1 p.G411S suggests that the p.G411S mutant PINK1 poisons the wild-type PINK1 so that it adopts the same misalignment of ubiquitin and ATP at the active site, impairing kinase function. While the conclusions from the 3D modelling must be interpreted cautiously without experimental structural data, this nonetheless represents key evidence that through dimerization p.G411S may exert a dominant negative effect.
Finally the authors confirmed that wild-type and p.G411S can dimerize when overexpressed in Hela cells, and that the heterodimer displays reduced kinase activity with respect to wild-type homodimers. Overexpression of wild-type PINK1 in this model is able to activate parkin translocation to the mitochondria. However co-expression of p.G411S (but not Q456X) with wild-type PINK1 significantly reduces mitochondrial translocation of parkin, confirming that the dominant negative effect of the p.G411S variant in reducing PINK1 kinase function also translates into downstream impairment of parkin recruitment.
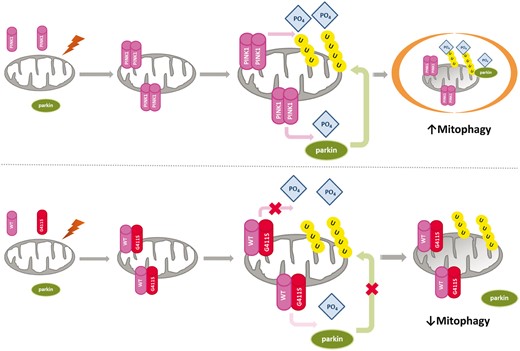
Putative mechanism of p.G411S variant.Top: Function of wild-type PINK1. Damage to mitochondria results in the formation of PINK1 homodimers at the mitochondria. The kinase function of PINK1 induces ubiquitin phosphorylation, as well as phosphorylation and activation of parkin, which is translocated to the mitochondria, culminating in engulfment of damaged mitochondria by autophagosomes and degradation by lysosomes. Bottom: The dominant negative mechanism of the heterozygous PINK1 p.G411S mutation. Damage to mitochondria results in the formation of dimers at the mitochondria that may be heterodimers consisting of wild-type PINK1 and G411S PINK1. The mutation confers a dominant negative effect disrupting the normal kinase function of the wild-type PINK1, leading to reduced ubiquitin phosphorylation, and reduced parkin activation and translocation. Ultimately this would lead to impaired mitophagy and accumulation of damaged mitochondria. WT = wild-type.
Glossary
Dominant negative: A single mutant allele exerts a negative effect on the normal DNA, RNA or protein resulting in impaired protein function
Haploinsufficiency: A single mutant allele results in 50% activity of the encoded protein, which is insufficient to maintain normal function
Mitophagy: The selective targeting and elimination of defective mitochondria by autophagy in order to avoid the accumulation of toxic mitochondria PINK1: PTEN-induced kinase 1
Ubiquitin kinase: An enzyme with the ability to transfer phosphate groups to the protein ubiquitin


{kind=link}