




Abstract
Hereditary spastic paraplegias are heterogeneous neurological disorders characterized by a pyramidal syndrome with symptoms predominantly affecting the lower limbs. Some limited pyramidal involvement also occurs in patients with an autosomal recessive neurocutaneous syndrome due to ALDH18A1 mutations. ALDH18A1 encodes delta-1-pyrroline-5-carboxylate synthase (P5CS), an enzyme that catalyses the first and common step of proline and ornithine biosynthesis from glutamate. Through exome sequencing and candidate gene screening, we report two families with autosomal recessive transmission of ALDH18A1 mutations, and predominant complex hereditary spastic paraplegia with marked cognitive impairment, without any cutaneous abnormality. More interestingly, we also identified monoallelic ALDH18A1 mutations segregating in three independent families with autosomal dominant pure or complex hereditary spastic paraplegia, as well as in two sporadic patients. Low levels of plasma ornithine, citrulline, arginine and proline in four individuals from two families suggested P5CS deficiency. Glutamine loading tests in two fibroblast cultures from two related affected subjects confirmed a metabolic block at the level of P5CS in vivo. Besides expanding the clinical spectrum of ALDH18A1-related pathology, we describe mutations segregating in an autosomal dominant pattern. The latter are associated with a potential trait biomarker; we therefore suggest including amino acid chromatography in the clinico-genetic work-up of hereditary spastic paraplegia, particularly in dominant cases, as the associated phenotype is not distinct from other causative genes.
Mutations in ALDH18A1, encoding an enzyme involved in glutamate metabolism, have been implicated in an autosomal recessive neurocutaneous disorder. Coutelier et al. widen the associated phenotype to include spastic paraplegia without cutaneous signs. They also report autosomal dominant transmission of ALDH18A1 mutations, associated with abnormal levels of plasma amino acids.
Introduction
Hereditary spastic paraplegias (HSPs) are a family of neurodegenerative disorders mainly affecting the corticospinal tract. Clinically, they are characterized by a pyramidal syndrome with paraparesis and spasticity, primarily detectable in the lower limbs and in the gait, and classified as either pure or complex, according to the absence or presence of accompanying neurological or extra-neurological signs (Blackstone, 2012; Finsterer et al., 2012). Genetically, they can follow all modes of inheritance: autosomal dominant, autosomal recessive, X-linked and mitochondrial. Many genes have already been described, and more are regularly identified (Novarino et al., 2014); in this respect, next generation sequencing techniques are powerful tools to elucidate the genetic basis of HSPs, either for known gene screening or for new gene identification.
In the last decade, mutations in ALDH18A1 have been implicated in an autosomal recessive neurocutaneous syndrome, characterized by severe developmental delay with marked cognitive impairment, associated with progeroid features, cutis laxa, joint hyperlaxity, short stature, cataract and frequent microcephaly (Baumgartner et al., 2000, 2005; Bicknell et al., 2008; Skidmore et al., 2011; Martinelli et al., 2012; Zampatti et al., 2012; Fischer et al., 2014; Gardeitchik et al., 2014; Handley et al., 2014; Wolthuis et al., 2014). Pyramidal signs were reported in 12 of 15 patients identified so far, but were often limited to brisk deep tendon reflexes. ALDH18A1 encodes delta-1-pyrroline-5-carboxylate synthase (P5CS), an enzyme that catalyses the first common steps of proline and ornithine biosynthesis from glutamate (Hu et al., 1999). Plasma amino acid levels were abnormal in three reported families (Baumgartner et al., 2000, 2005; Martinelli et al., 2012; Fischer et al., 2014), with low proline, ornithine (2/3), citrulline and arginine (3/3) levels associated with mild fasting hyperammonaemia. In this paper, we report seven pedigrees with ALDH18A1 mutations segregating in a recessive or, for the first time, a dominant inheritance mode associated with abnormal plasma amino acid levels.
Materials and methods
Patient recruitment and clinical evaluation
Families FSP410, FSP429, FSP470, FSP856 and SR45 were of French (n = 2), Spanish (n = 1), Italian (n = 1) or Portuguese (n = 1) ancestry and had been identified as part of the SPATAX (https://spatax.wordpress.com/) cohort of patients with HSP. Two sporadic cases were Caucasian, from Australia/UK and USA. All patients were examined by at least one of the co-authors. Blood samples were obtained after informed and signed consent according to local ethics regulations. DNA was extracted using a standard protocol.
Whole genome mapping and exome sequencing in Family FSP410
Whole genome linkage analysis was performed using Illumina LINKAGE_12 SNP microarrays. Genotypes were determined using Beadstudio (Illumina) and analysed with MERLIN 1.0 (Abecasis et al., 2002) (Supplementary Fig. 1). Exome sequencing was performed on four affected subjects (Patients 6, 8, 25 and 29; Fig. 1). Targeted capture was done with the Agilent SureSelect Human All Exon Capture V4 XT 51MB kit. Paired-end 100 bp sequencing was performed on an Illumina HiSeq2000 system. Sequence reads were trimmed using FastX and duplicate reads were removed by Picard v1.59 (http://picard.sourceforge.net). Sequence reads were aligned to the human genome reference (hg19) using the Burroughs-Wheeler algorithm v0.6.2, followed by a local realignment around indels (insertions/deletions) using the Broad Institute Genome Analysis Tool Kit (GATK v1.4; DePristo et al., 2011). Variants (SNP/INDEL) were called using GATK's Unified Genotyper module and annotated with Annovar (Wang et al., 2010). They were sorted according to their localization in putatively linked or not excluded loci, expected transmission mode (heterozygous in all four individuals), frequency lower than 0.1% in public databases (CompleteGenomics, http://www.completegenomics.com/public-data/69-Genomes/; EVS, http://evs.gs.washington.edu/EVS; 1000genomes, http://www.1000genomes.org/; Exome Aggregation Consortium, http://exac.broadinstitute.org/), absence in internal controls, frequency in internal database (<5%), effect on the coding sequence (non-synonymous, splice site, frameshift or nonsense), coverage above 10×, and amino acid conservation score (GERP++ > 0, (http://mendel.stanford.edu/SidowLab/downloads/gerp/). Segregation in all affected individuals was checked through Sanger sequencing according to standard protocols.
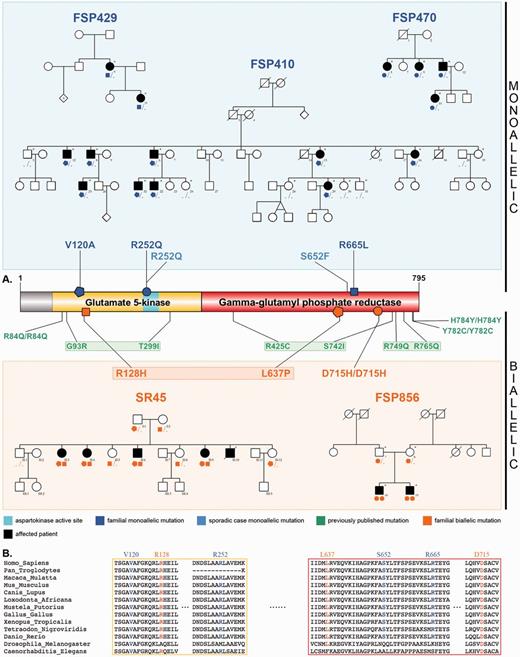
Pedigrees, ALDH18A1 mutations and conservation across species. (A) Schematics of P5CS and its gamma-glutamyl kinase and gamma-glutamyl phosphate reductase domains. Newly described monoallelic mutations (blue), newly described biallelic mutations (orange), and previously reported biallelic mutations (green) are represented, along with pedigrees of the families of this study, showing the segregation. Sampled individuals are indicated with an asterisk. In autosomal dominant families, all affected patients carry one ALDH18A1 mutation; the penetrance seems to be complete as no tested unaffected family member has any mutation. (B) Alignment of P5CS paralogues, showing the high conservation of all affected residues reported.
ALDH18A1 analysis in 435 exomes of index patients with hereditary spastic paraplegia
To assess the frequency of ALDH18A1 mutations in autosomal dominant HSP, exome sequencing data of 160 index patients with autosomal dominant HSP were examined. Because of the previous description of ALDH18A1 mutations transmitted in an autosomal recessive mode of inheritance, the search was extended to 275 index patients with autosomal recessive or sporadic HSP. Exome data were shared and are available (GEM.app database, https://genomics.med.miami.edu/) as part of an international collaborative effort (Gonzalez et al., 2013).
Panel sequencing in 95 index patients with hereditary spastic paraplegia
An amplicon-based panel including all exons of ALDH18A1 was designed in-house with Primer3Plus (Supplementary Table 1). For 95 patients with autosomal dominant and recessive HSP selected from the SPATAX cohort, mostly with either European (32/45 with available origin) or North African (12/45) ancestry, all amplicons were amplified using the Fluidigm Access Array technology (IFC Controller AX, FC1 Cycler, 48.48 Access Arrays), and sequenced on the MiSeq Illumina sequencer as paired-end 2 × 250 bp reads. The Burrows-Wheeler algorithm v0.7.8 was applied to align sequence reads to the UCSC Genome Browser hg19 version of the human genome and variants were called via the GATK software package v3.1‐1 after realignment and recalibration. Variants meeting the aforementioned criteria were confirmed using Sanger sequencing, and segregation in other affected members in the family was verified, when possible. In all ALDH18A1-mutated index cases, mutations in 74 previously described HSP genes were excluded with a Roche/Nimblegen capture in-house panel followed by MiSeq sequencing (unpublished data).
Biochemical analyses
Plasma amino acid profiles were obtained by ion-exchange chromatography with standard ninhydrin detection (JEOL Aminotac JLC 500/V) as part of routine diagnostic work-up for Patients FSP410‐13, -29, -32, FSP429‐21, and FSP856‐18 on blood and urine samples. The results were compared with those obtained from the first family published with ALDH18A1 mutations (Baumgartner et al., 2005) and from the full cohort of patients from the Necker Hospital (Paris) between 1995 and 2013 (n > 53 000), after exclusion of patients <15 years or with a known metabolic disorder, leading to a final set of 5023 patients. Age normalization was performed by locally weighted regression (loess) on a reference hospital population of 5043 individuals (data not shown).
Dermal fibroblasts from Patients FSP410‐29 and FSP410‐32 were grown in Dulbecco’s modified Eagle’s medium with 10% foetal bovine serum and 1% penicillin-streptomycin in a 37°C incubator with 5% CO2. After removing the culture medium, they were incubated with 2.5 mM glucose and 1 mM 13C5-stable isotope labelled glutamine in phosphate buffer for 18 h. Incubation was quenched by methanol, samples were silylated [N,O-bis(trimethylsilyl)trifluoroacetamide + 1% trimethylchlorosilane] and analysed by gas chromatography coupled to a triple quadrupole mass spectrometer (Scion, Brüker). Quantification of labelled versus natural isotope ions was performed in selected reaction monitoring mode.
Results
Identification of a mutation in ALDH18A1 in Family FSP410
Under an autosomal dominant inheritance model, whole genome linkage analysis in Family FSP410 identified five putatively linked loci with multipoint logarithm of odds (LOD) scores reaching the maximal expected values for this pedigree (ranging from +1.64 to +1.95) as well as various uninformative regions with LOD scores varying from −1.90 to +0.70 (Supplementary Fig. 1). Whole exome sequencing performed in four patients provided 117 to 130 million reads per sample, 98% of which could be aligned to the targeted sequence. Mean depth of the targeted sequence was 125- to 132-fold. From 114 285 to 119 137 SNPs (single nucleotide polymorphisms) and from 10 546 to 11 159 indels were identified. Two missense variants respecting the abovementioned criteria segregated with the disease in the entire family: a c.359T > C/p.V120A (NM_002860.3) change in ALDH18A1 (OMIM 138250), at chr10:g.97 397 138; and a c.2250G > T/p.W750C (NM_001195263) change in PDZD7 (OMIM 612971), at chr10:g.102 770 396. Only the ALDH18A1 variant was predicted to be deleterious by all four prediction software used (Table 1) and was located in a functional domain of the protein. It affected a highly conserved amino acid across species (Fig. 1). ALDH18A1 was previously implicated in an autosomal recessive syndrome that associates cutaneous, ocular, metabolic and neurological findings, with pyramidal signs (Baumgartner et al., 2005), often limited to brisk reflexes; PDZD7 contributes to Usher syndrome type II (OMIM 605472), either as a modifier or in a digenic pattern of transmission (Ebermann et al., 2010). We found no additional causative PDZD7 variants in 95 HSP index cases and had less in silico elements in favour of its pathogenicity; we therefore focused on ALDH18A1.
Characteristics of novel ALDH18A1 mutations
| Mutation characteristics | Database frequency | Pathogenicity prediction | Conservation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | hg19 (chr10) | cDNA and protein changes | dbSNP 138 | EVS | ExAC | SIFT score | PolyPhen-2 HDIV | PolyPhen-2 HVAR | LRT score | MutationTaster score | GERP++ score | PhyloP score |
| FSP410 | 97397138 | NM_002860.3: c.359T > C; p.Val120Ala | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.995 (D) | D | D (0.999993) | 5.6 | 2.134 |
| FSP429 | 97371129 | NM_002860.3: c.1994G > T; p.Arg665Leu | 0 | 0 | 0/122000+ | 0.06 | 0.949 (P) | 0.78 (P) | D | D (0.999969) | 5.6 | 2.639 |
| FSP470 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| GSHSP44 | 97371168 | NM_002860.3: c.1955C > T; p.Ser652Phe | 0 | 0 | 0/122000+ | 0.02 (D) | 0.892 (P) | 0.694 (P) | D | D (0.999952) | 5.6 | 2.639 |
| 25014 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| FSP856 | 97370017 | NM_002860.3: c.2143G > C; p.Asp715His | 0 | 0 | 1/122892 - 0 hmz | 0 (D) | 1 (D) | 1 (D) | D | D (0.999999) | 5.43 | 2.722 |
| SR45 – mutation 1 | 97397114 | NM_002860.3: c.383G > A; p.Arg128His | 0 | 0 | 4/122890 - 1 hmz | 0.01 (D) | 0.999 (D) | 0.978 (D) | D | D (0.999981) | 5.6 | 2.642 |
| SR45 – mutation 2 | 97373512 | NM_002860.3: c.1910T > C; p.Leu637Pro | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.999 (D) | D | D (0.999999) | 6.07 | 2.326 |
| Mutation characteristics | Database frequency | Pathogenicity prediction | Conservation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | hg19 (chr10) | cDNA and protein changes | dbSNP 138 | EVS | ExAC | SIFT score | PolyPhen-2 HDIV | PolyPhen-2 HVAR | LRT score | MutationTaster score | GERP++ score | PhyloP score |
| FSP410 | 97397138 | NM_002860.3: c.359T > C; p.Val120Ala | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.995 (D) | D | D (0.999993) | 5.6 | 2.134 |
| FSP429 | 97371129 | NM_002860.3: c.1994G > T; p.Arg665Leu | 0 | 0 | 0/122000+ | 0.06 | 0.949 (P) | 0.78 (P) | D | D (0.999969) | 5.6 | 2.639 |
| FSP470 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| GSHSP44 | 97371168 | NM_002860.3: c.1955C > T; p.Ser652Phe | 0 | 0 | 0/122000+ | 0.02 (D) | 0.892 (P) | 0.694 (P) | D | D (0.999952) | 5.6 | 2.639 |
| 25014 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| FSP856 | 97370017 | NM_002860.3: c.2143G > C; p.Asp715His | 0 | 0 | 1/122892 - 0 hmz | 0 (D) | 1 (D) | 1 (D) | D | D (0.999999) | 5.43 | 2.722 |
| SR45 – mutation 1 | 97397114 | NM_002860.3: c.383G > A; p.Arg128His | 0 | 0 | 4/122890 - 1 hmz | 0.01 (D) | 0.999 (D) | 0.978 (D) | D | D (0.999981) | 5.6 | 2.642 |
| SR45 – mutation 2 | 97373512 | NM_002860.3: c.1910T > C; p.Leu637Pro | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.999 (D) | D | D (0.999999) | 6.07 | 2.326 |
Summary of characteristics for all variants reported. The database frequencies were looked for in dbSNP138 (http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS) and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/). Pathogenicity scores are evaluated as follows: SIFT (http://sift.jcvi.org/) predicts deleteriousness (D) under 0.05; PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) classifies SNPs as probably damaging (D; HDIV ≥ 0.957, HVAR ≥ 0.909), possibly damaging (P; 0.453 ≤ HDIV ≤ 0.956, 0.447 ≤ pp2_hdiv ≤ 0.908), or benign (B; HDIV ≤ 0.452, HVAR ≤ 0.446); LRT (Chun and Fay, 2009) differentiates variants with deleterious (D), neutral (N) or unknown (U) effect; MutationTaster (http://www.mutationtaster.org/) classifies them as ‘disease_causing_automatic’ (A), ‘disease_causing’ (D), ‘polymorphism’ (N) or ‘polymorphism_automatic’ (P) with a given probability value, 1 being the most probable. For both GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/) and PhyloP (http://compgen.bscb.cornell.edu/phast/help-pages/phyloP.txt), higher scores indicate better residue conservation.
Hmz = homozygous.
Characteristics of novel ALDH18A1 mutations
| Mutation characteristics | Database frequency | Pathogenicity prediction | Conservation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | hg19 (chr10) | cDNA and protein changes | dbSNP 138 | EVS | ExAC | SIFT score | PolyPhen-2 HDIV | PolyPhen-2 HVAR | LRT score | MutationTaster score | GERP++ score | PhyloP score |
| FSP410 | 97397138 | NM_002860.3: c.359T > C; p.Val120Ala | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.995 (D) | D | D (0.999993) | 5.6 | 2.134 |
| FSP429 | 97371129 | NM_002860.3: c.1994G > T; p.Arg665Leu | 0 | 0 | 0/122000+ | 0.06 | 0.949 (P) | 0.78 (P) | D | D (0.999969) | 5.6 | 2.639 |
| FSP470 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| GSHSP44 | 97371168 | NM_002860.3: c.1955C > T; p.Ser652Phe | 0 | 0 | 0/122000+ | 0.02 (D) | 0.892 (P) | 0.694 (P) | D | D (0.999952) | 5.6 | 2.639 |
| 25014 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| FSP856 | 97370017 | NM_002860.3: c.2143G > C; p.Asp715His | 0 | 0 | 1/122892 - 0 hmz | 0 (D) | 1 (D) | 1 (D) | D | D (0.999999) | 5.43 | 2.722 |
| SR45 – mutation 1 | 97397114 | NM_002860.3: c.383G > A; p.Arg128His | 0 | 0 | 4/122890 - 1 hmz | 0.01 (D) | 0.999 (D) | 0.978 (D) | D | D (0.999981) | 5.6 | 2.642 |
| SR45 – mutation 2 | 97373512 | NM_002860.3: c.1910T > C; p.Leu637Pro | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.999 (D) | D | D (0.999999) | 6.07 | 2.326 |
| Mutation characteristics | Database frequency | Pathogenicity prediction | Conservation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | hg19 (chr10) | cDNA and protein changes | dbSNP 138 | EVS | ExAC | SIFT score | PolyPhen-2 HDIV | PolyPhen-2 HVAR | LRT score | MutationTaster score | GERP++ score | PhyloP score |
| FSP410 | 97397138 | NM_002860.3: c.359T > C; p.Val120Ala | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.995 (D) | D | D (0.999993) | 5.6 | 2.134 |
| FSP429 | 97371129 | NM_002860.3: c.1994G > T; p.Arg665Leu | 0 | 0 | 0/122000+ | 0.06 | 0.949 (P) | 0.78 (P) | D | D (0.999969) | 5.6 | 2.639 |
| FSP470 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| GSHSP44 | 97371168 | NM_002860.3: c.1955C > T; p.Ser652Phe | 0 | 0 | 0/122000+ | 0.02 (D) | 0.892 (P) | 0.694 (P) | D | D (0.999952) | 5.6 | 2.639 |
| 25014 | 97392769 | NM_002860.3: c.755G > A; p.Arg252Gln | 0 | 0 | 0/122000+ | 0.23 | 0.997 (D) | 0.941 (D) | D | D (0.999779) | 5.97 | 2.832 |
| FSP856 | 97370017 | NM_002860.3: c.2143G > C; p.Asp715His | 0 | 0 | 1/122892 - 0 hmz | 0 (D) | 1 (D) | 1 (D) | D | D (0.999999) | 5.43 | 2.722 |
| SR45 – mutation 1 | 97397114 | NM_002860.3: c.383G > A; p.Arg128His | 0 | 0 | 4/122890 - 1 hmz | 0.01 (D) | 0.999 (D) | 0.978 (D) | D | D (0.999981) | 5.6 | 2.642 |
| SR45 – mutation 2 | 97373512 | NM_002860.3: c.1910T > C; p.Leu637Pro | 0 | 0 | 0/122000+ | 0 (D) | 1 (D) | 0.999 (D) | D | D (0.999999) | 6.07 | 2.326 |
Summary of characteristics for all variants reported. The database frequencies were looked for in dbSNP138 (http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS) and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/). Pathogenicity scores are evaluated as follows: SIFT (http://sift.jcvi.org/) predicts deleteriousness (D) under 0.05; PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) classifies SNPs as probably damaging (D; HDIV ≥ 0.957, HVAR ≥ 0.909), possibly damaging (P; 0.453 ≤ HDIV ≤ 0.956, 0.447 ≤ pp2_hdiv ≤ 0.908), or benign (B; HDIV ≤ 0.452, HVAR ≤ 0.446); LRT (Chun and Fay, 2009) differentiates variants with deleterious (D), neutral (N) or unknown (U) effect; MutationTaster (http://www.mutationtaster.org/) classifies them as ‘disease_causing_automatic’ (A), ‘disease_causing’ (D), ‘polymorphism’ (N) or ‘polymorphism_automatic’ (P) with a given probability value, 1 being the most probable. For both GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/) and PhyloP (http://compgen.bscb.cornell.edu/phast/help-pages/phyloP.txt), higher scores indicate better residue conservation.
Hmz = homozygous.
Additional families with ALDH18A1 mutations
To confirm the implication of ALDH18A1 in autosomal dominant HSP, and further assess its potential role in autosomal recessive HSP, we screened 530 patients through whole exome sequencing (n = 435) or panel sequencing (n = 95). Segregating heterozygous variants were found in two French families compatible with an autosomal dominant inheritance mode: a c.1994G > T/p.R665L change at g.97 371 129 in Family FSP429, and a c.755G > A/p.R252Q change at g.97 392 769 in Family FSP470. The same p.R252Q change was also identified in Patient 25014 with pure HSP living in the USA. Reference haplotypes for two SNPs flanking the ALDH18A1 mutation (rs 9787589, rs 10882640) allowed excluding the existence of a common ancestor with Family FSP470, that presents homozygous variants at both positions. A heterozygous c.1949G > T/p.S652F change at g.97 371 198 was identified in a sporadic patient from Australia/UK (Patient GSHSP44). In two families with complex autosomal recessive HSP, biallelic variants were identified: compound heterozygosity for a c.1910T > C/p.L637P change at g.97 373 512 and a c.383G > A/p.R128H change at g.97 397 114 in Family SR45 from Portugal, and a homozygous c.2143G > C/p.D715H change at g.97 370 017 in Spanish Family FSP856. All mutations affected conserved amino acids and were absent or rare in exome databases (Fig. 1 and Table 1).
Clinical characteristics of patients with ALDH18A1 mutations
In Family FSP410, clinical assessment was available for seven of nine patients (Table 2). The common clinical picture was a complex, slowly progressive but finally severe HSP, with onset in adolescence or adulthood (ranging from 14 to 59 years) and predominant gait signs, motor neuropathy (3/4 tested) and spastic dysarthria (4/6). Mild cerebellar signs in gait (3/6) and upper limbs (1/6) were described; one patient presented with dementia at age 69. One carrier (Patient FSP410‐22) presented with congenital cataract but also showed, on last examination at age 28, an isolated mild pyramidal syndrome of the four limbs with enhanced upper limbs reflexes, brisk patellar reflexes, absent ankle reflexes, bilateral Hoffman signs and unilaterally indifferent plantar cutaneous reflex with no spasticity. Families FSP429 and FSP470 presented with moderate to severe spasticity in gait with few or no associated signs. Cerebellar signs were present in Family FSP429 (1/2), cataracts and gastrointestinal reflux in Patient FSP470‐22. Brain MRI was abnormal in Patient FSP429‐6 with mild FLAIR hyperintensity of the corticospinal tract posterior of the ventricles, a localized and heterogeneous T2 hyperintensity in the pons, and moderate spinal cord atrophy (Supplementary Fig. 2). Both additional heterozygous cases presented with pure HSP, with spinal cord atrophy on MRI in Patient GSHSP44. Pes cavus was reported in 7 of 15 patients with monoallelic ALDH18A1 variant.
Clinical characteristics of patients with ALDH18A1 mutations: autosomal dominant families
| Family N°/origin | FSP410/Italy | FSP429/France | FSP470/France | Spo/USA | Spo/Australia and UK | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH18A1 variant/inheritance | p.Val120Ala/Autosomal dominant | p.Arg665Leu/Dominant | p.Arg252Gln/Dominant | p.Arg252Gln/Sporadic | p.Ser652Phe/Sporadic | ||||||||||
| Individual N° (sex) | 29 (F) | 13 (F) | 6 (M) | 32 (M) | 8 (M) | 16 (F) | 22 (M) | 6 (F) | 21 (F) | 5 (F) | 9 (F) | 11 (M) | 22 (F) | 25014 (F) | GSHSP44 (F) |
| Age at exam (years) | 45 | 70 | 71 | 43 | 61 | 50 | 28 | 66 | 37 | 52 | 50 | 48 | 20 | 76 | 46 |
| Age at onset (years) | 18 | 22 | 49 | 14 | 59 | 42 | ? | 24 | 13 | 19 | 21 | 43 | 1 | 31 | 44 |
| Symptoms at onset | Muscle cramps | Weakness | Stiff legs | Stiff legs | Pain and weakness in LL | Stiff legs, mild instability | Reflex pyramidal syndrome | Stiff legs | Unsteadiness | Falls | Stiff legs | Stiff legs | Stiff legs | Falls | Toe walking, tripping |
| Disease duration (years) | 27 | 48 | 22 | 29 | 2 | 25 | ? | 34 | 24 | 33 | 29 | 5 | 19 | 45 | 12 |
| Disability score (max 7) | 4/7 | 5/7 | 7/7 | 3/7 | NA | 3/7 | 0/7 | 6/7 | 4/7 | 3/7 | 4/7 | 4/7 | 3/7 | 5/7 | 4/7 |
| Spasticity at gait | Severe | Severe | Severe | Moderate | NA | Moderate | None | Severe | Severe | Moderate | Severe | Moderate | Moderate | Moderate | Mild |
| Spasticity at rest | Mild Ashworth 1/4 | Mild | Severe | Mild | NA | Mild | None | None Ashworth 0/4 | Moderate | Moderate Ashworth 2/4 | Mild Ashworth 2/4 | Mild | Mild | Severe | Mild |
| Weakness | Distal > proximal | Diffuse (3/5 MRC grading 4 limbs) | Proximal, distal | Distal > proximal (4 limbs) | L > R | Distal (3/5) > proximal | None | Distal | Distal > proximal | Proximal, distal | Proximal, distal | Proximal, distal | Proximal, distal | Distal > proximal | Distal > proximal |
| Increased reflexes LL | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes | Yes | Yes | Yes (ankles decreased) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Normal | Yes | Yes | Yes | Yes | Yes | Yes |
| Extensor plantar response | Indifferent | Yes | Yes | Indifferent | Yes (R) | Yes | R indifferent | Yes | Indifferent | Yes | Yes | Yes | Yes | Yes | Yes |
| Hoffman sign | NA | NA | NA | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | No | NA | NA |
| Decreased vibration sense at ankles | ND | ND | Yes | Mild | NA | No | No | No | Mild | No | No | No | No | Moderate | No |
| Urinary symptoms | Pollakiury | No | Yes | Pollakiury | NA | Yes | No | No | Urinary incontinence | Urinary urgency | Urinary urgency | No | No | Urinary urgency | Urinary urgency |
| Cerebellar gait | No | No | Mild | Mild | NA | Mild | No | Yes (CCFS 0.939) | No | No | No | No | No | No | No |
| Cerebellar signs UL | No | No | Mild | No | NA | No | No | Yes | No | No | No | No | No | No | No |
| Dysarthria | Spastic | No | Anarthria (age 70) | Spastic | NA | Spastic | No | No | Yes | No | No | No | No | No | No |
| Cognitive impairment | No | No | Dementia (age 69) | No | Mild memory impairment | Mild memory impairment | No | No | No | No | No | No | No | No | No |
| Cutaneous findings | No | No | No | No | NA | No | No | No | No | No | No | No | No | Chronic cellulitis changes | No |
| Ocular findings | NA | NA | NA | NA | NA | NA | Congenital cataract | NA | NA | NA | NA | NA | Cataract (age 8 months) | Bilateral senile cataracts (69) | NA |
| Other clinical features | Pes cavus, neuropathic pain | Neuropathic pain | Low back pain, neuropathic pain (LL) | Pes cavus, infantile psychosis, mild horizontal nystagmus | Neuropathic pain (LL), gaze nystagmus, neurosensory deafness, temporal epilepsy | Mild pes cavus | Pes cavus | Pain (knee and back) | Pes cavus | Pes cavus, tremor (left UL) | Mitral leak | Gastrointestinal reflux | Resting tremor, uterine cancer (69) | Pes cavus, hip replacement, groin pain | |
| MRI | Normal brain MRI (2002) | ND | ND | Mild CC atrophy | Normal brain MRI | Large cisterna magna on MRI | NA | White matter anomalies, pons hypersignal, thin dorsal cord | Thin dorsal cord | NA | Left arachnoidien cyst | NA | NA | Cortical atrophy (frontoparietal predominance), chronic microvascular disease (69) | Spinal cord atrophy |
| Other paraclinical tests | ENMG: motor neuronopathy (2009) | NA | NA | ENMG: motor neuronopathy (2014) | Normal ENMG | ENMG: motor neuronopathy (LL, L>R) (2004) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Family N°/origin | FSP410/Italy | FSP429/France | FSP470/France | Spo/USA | Spo/Australia and UK | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH18A1 variant/inheritance | p.Val120Ala/Autosomal dominant | p.Arg665Leu/Dominant | p.Arg252Gln/Dominant | p.Arg252Gln/Sporadic | p.Ser652Phe/Sporadic | ||||||||||
| Individual N° (sex) | 29 (F) | 13 (F) | 6 (M) | 32 (M) | 8 (M) | 16 (F) | 22 (M) | 6 (F) | 21 (F) | 5 (F) | 9 (F) | 11 (M) | 22 (F) | 25014 (F) | GSHSP44 (F) |
| Age at exam (years) | 45 | 70 | 71 | 43 | 61 | 50 | 28 | 66 | 37 | 52 | 50 | 48 | 20 | 76 | 46 |
| Age at onset (years) | 18 | 22 | 49 | 14 | 59 | 42 | ? | 24 | 13 | 19 | 21 | 43 | 1 | 31 | 44 |
| Symptoms at onset | Muscle cramps | Weakness | Stiff legs | Stiff legs | Pain and weakness in LL | Stiff legs, mild instability | Reflex pyramidal syndrome | Stiff legs | Unsteadiness | Falls | Stiff legs | Stiff legs | Stiff legs | Falls | Toe walking, tripping |
| Disease duration (years) | 27 | 48 | 22 | 29 | 2 | 25 | ? | 34 | 24 | 33 | 29 | 5 | 19 | 45 | 12 |
| Disability score (max 7) | 4/7 | 5/7 | 7/7 | 3/7 | NA | 3/7 | 0/7 | 6/7 | 4/7 | 3/7 | 4/7 | 4/7 | 3/7 | 5/7 | 4/7 |
| Spasticity at gait | Severe | Severe | Severe | Moderate | NA | Moderate | None | Severe | Severe | Moderate | Severe | Moderate | Moderate | Moderate | Mild |
| Spasticity at rest | Mild Ashworth 1/4 | Mild | Severe | Mild | NA | Mild | None | None Ashworth 0/4 | Moderate | Moderate Ashworth 2/4 | Mild Ashworth 2/4 | Mild | Mild | Severe | Mild |
| Weakness | Distal > proximal | Diffuse (3/5 MRC grading 4 limbs) | Proximal, distal | Distal > proximal (4 limbs) | L > R | Distal (3/5) > proximal | None | Distal | Distal > proximal | Proximal, distal | Proximal, distal | Proximal, distal | Proximal, distal | Distal > proximal | Distal > proximal |
| Increased reflexes LL | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes | Yes | Yes | Yes (ankles decreased) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Normal | Yes | Yes | Yes | Yes | Yes | Yes |
| Extensor plantar response | Indifferent | Yes | Yes | Indifferent | Yes (R) | Yes | R indifferent | Yes | Indifferent | Yes | Yes | Yes | Yes | Yes | Yes |
| Hoffman sign | NA | NA | NA | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | No | NA | NA |
| Decreased vibration sense at ankles | ND | ND | Yes | Mild | NA | No | No | No | Mild | No | No | No | No | Moderate | No |
| Urinary symptoms | Pollakiury | No | Yes | Pollakiury | NA | Yes | No | No | Urinary incontinence | Urinary urgency | Urinary urgency | No | No | Urinary urgency | Urinary urgency |
| Cerebellar gait | No | No | Mild | Mild | NA | Mild | No | Yes (CCFS 0.939) | No | No | No | No | No | No | No |
| Cerebellar signs UL | No | No | Mild | No | NA | No | No | Yes | No | No | No | No | No | No | No |
| Dysarthria | Spastic | No | Anarthria (age 70) | Spastic | NA | Spastic | No | No | Yes | No | No | No | No | No | No |
| Cognitive impairment | No | No | Dementia (age 69) | No | Mild memory impairment | Mild memory impairment | No | No | No | No | No | No | No | No | No |
| Cutaneous findings | No | No | No | No | NA | No | No | No | No | No | No | No | No | Chronic cellulitis changes | No |
| Ocular findings | NA | NA | NA | NA | NA | NA | Congenital cataract | NA | NA | NA | NA | NA | Cataract (age 8 months) | Bilateral senile cataracts (69) | NA |
| Other clinical features | Pes cavus, neuropathic pain | Neuropathic pain | Low back pain, neuropathic pain (LL) | Pes cavus, infantile psychosis, mild horizontal nystagmus | Neuropathic pain (LL), gaze nystagmus, neurosensory deafness, temporal epilepsy | Mild pes cavus | Pes cavus | Pain (knee and back) | Pes cavus | Pes cavus, tremor (left UL) | Mitral leak | Gastrointestinal reflux | Resting tremor, uterine cancer (69) | Pes cavus, hip replacement, groin pain | |
| MRI | Normal brain MRI (2002) | ND | ND | Mild CC atrophy | Normal brain MRI | Large cisterna magna on MRI | NA | White matter anomalies, pons hypersignal, thin dorsal cord | Thin dorsal cord | NA | Left arachnoidien cyst | NA | NA | Cortical atrophy (frontoparietal predominance), chronic microvascular disease (69) | Spinal cord atrophy |
| Other paraclinical tests | ENMG: motor neuronopathy (2009) | NA | NA | ENMG: motor neuronopathy (2014) | Normal ENMG | ENMG: motor neuronopathy (LL, L>R) (2004) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
CC = corpus callosum; CCFS = composite cerebellar functional severity score; ENMG = electro-neuro-myogram; HC = head circumference; ID = intellectual delay; L = left; LL = lower limbs; MD = motor delay; MRC = medical research council; NA = not available; ND = not done; PMD = psychomotor delay; R = right; UL = upper limbs.
Clinical characteristics of patients with ALDH18A1 mutations: autosomal dominant families
| Family N°/origin | FSP410/Italy | FSP429/France | FSP470/France | Spo/USA | Spo/Australia and UK | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH18A1 variant/inheritance | p.Val120Ala/Autosomal dominant | p.Arg665Leu/Dominant | p.Arg252Gln/Dominant | p.Arg252Gln/Sporadic | p.Ser652Phe/Sporadic | ||||||||||
| Individual N° (sex) | 29 (F) | 13 (F) | 6 (M) | 32 (M) | 8 (M) | 16 (F) | 22 (M) | 6 (F) | 21 (F) | 5 (F) | 9 (F) | 11 (M) | 22 (F) | 25014 (F) | GSHSP44 (F) |
| Age at exam (years) | 45 | 70 | 71 | 43 | 61 | 50 | 28 | 66 | 37 | 52 | 50 | 48 | 20 | 76 | 46 |
| Age at onset (years) | 18 | 22 | 49 | 14 | 59 | 42 | ? | 24 | 13 | 19 | 21 | 43 | 1 | 31 | 44 |
| Symptoms at onset | Muscle cramps | Weakness | Stiff legs | Stiff legs | Pain and weakness in LL | Stiff legs, mild instability | Reflex pyramidal syndrome | Stiff legs | Unsteadiness | Falls | Stiff legs | Stiff legs | Stiff legs | Falls | Toe walking, tripping |
| Disease duration (years) | 27 | 48 | 22 | 29 | 2 | 25 | ? | 34 | 24 | 33 | 29 | 5 | 19 | 45 | 12 |
| Disability score (max 7) | 4/7 | 5/7 | 7/7 | 3/7 | NA | 3/7 | 0/7 | 6/7 | 4/7 | 3/7 | 4/7 | 4/7 | 3/7 | 5/7 | 4/7 |
| Spasticity at gait | Severe | Severe | Severe | Moderate | NA | Moderate | None | Severe | Severe | Moderate | Severe | Moderate | Moderate | Moderate | Mild |
| Spasticity at rest | Mild Ashworth 1/4 | Mild | Severe | Mild | NA | Mild | None | None Ashworth 0/4 | Moderate | Moderate Ashworth 2/4 | Mild Ashworth 2/4 | Mild | Mild | Severe | Mild |
| Weakness | Distal > proximal | Diffuse (3/5 MRC grading 4 limbs) | Proximal, distal | Distal > proximal (4 limbs) | L > R | Distal (3/5) > proximal | None | Distal | Distal > proximal | Proximal, distal | Proximal, distal | Proximal, distal | Proximal, distal | Distal > proximal | Distal > proximal |
| Increased reflexes LL | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes | Yes | Yes | Yes (ankles decreased) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Normal | Yes | Yes | Yes | Yes | Yes | Yes |
| Extensor plantar response | Indifferent | Yes | Yes | Indifferent | Yes (R) | Yes | R indifferent | Yes | Indifferent | Yes | Yes | Yes | Yes | Yes | Yes |
| Hoffman sign | NA | NA | NA | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | No | NA | NA |
| Decreased vibration sense at ankles | ND | ND | Yes | Mild | NA | No | No | No | Mild | No | No | No | No | Moderate | No |
| Urinary symptoms | Pollakiury | No | Yes | Pollakiury | NA | Yes | No | No | Urinary incontinence | Urinary urgency | Urinary urgency | No | No | Urinary urgency | Urinary urgency |
| Cerebellar gait | No | No | Mild | Mild | NA | Mild | No | Yes (CCFS 0.939) | No | No | No | No | No | No | No |
| Cerebellar signs UL | No | No | Mild | No | NA | No | No | Yes | No | No | No | No | No | No | No |
| Dysarthria | Spastic | No | Anarthria (age 70) | Spastic | NA | Spastic | No | No | Yes | No | No | No | No | No | No |
| Cognitive impairment | No | No | Dementia (age 69) | No | Mild memory impairment | Mild memory impairment | No | No | No | No | No | No | No | No | No |
| Cutaneous findings | No | No | No | No | NA | No | No | No | No | No | No | No | No | Chronic cellulitis changes | No |
| Ocular findings | NA | NA | NA | NA | NA | NA | Congenital cataract | NA | NA | NA | NA | NA | Cataract (age 8 months) | Bilateral senile cataracts (69) | NA |
| Other clinical features | Pes cavus, neuropathic pain | Neuropathic pain | Low back pain, neuropathic pain (LL) | Pes cavus, infantile psychosis, mild horizontal nystagmus | Neuropathic pain (LL), gaze nystagmus, neurosensory deafness, temporal epilepsy | Mild pes cavus | Pes cavus | Pain (knee and back) | Pes cavus | Pes cavus, tremor (left UL) | Mitral leak | Gastrointestinal reflux | Resting tremor, uterine cancer (69) | Pes cavus, hip replacement, groin pain | |
| MRI | Normal brain MRI (2002) | ND | ND | Mild CC atrophy | Normal brain MRI | Large cisterna magna on MRI | NA | White matter anomalies, pons hypersignal, thin dorsal cord | Thin dorsal cord | NA | Left arachnoidien cyst | NA | NA | Cortical atrophy (frontoparietal predominance), chronic microvascular disease (69) | Spinal cord atrophy |
| Other paraclinical tests | ENMG: motor neuronopathy (2009) | NA | NA | ENMG: motor neuronopathy (2014) | Normal ENMG | ENMG: motor neuronopathy (LL, L>R) (2004) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Family N°/origin | FSP410/Italy | FSP429/France | FSP470/France | Spo/USA | Spo/Australia and UK | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH18A1 variant/inheritance | p.Val120Ala/Autosomal dominant | p.Arg665Leu/Dominant | p.Arg252Gln/Dominant | p.Arg252Gln/Sporadic | p.Ser652Phe/Sporadic | ||||||||||
| Individual N° (sex) | 29 (F) | 13 (F) | 6 (M) | 32 (M) | 8 (M) | 16 (F) | 22 (M) | 6 (F) | 21 (F) | 5 (F) | 9 (F) | 11 (M) | 22 (F) | 25014 (F) | GSHSP44 (F) |
| Age at exam (years) | 45 | 70 | 71 | 43 | 61 | 50 | 28 | 66 | 37 | 52 | 50 | 48 | 20 | 76 | 46 |
| Age at onset (years) | 18 | 22 | 49 | 14 | 59 | 42 | ? | 24 | 13 | 19 | 21 | 43 | 1 | 31 | 44 |
| Symptoms at onset | Muscle cramps | Weakness | Stiff legs | Stiff legs | Pain and weakness in LL | Stiff legs, mild instability | Reflex pyramidal syndrome | Stiff legs | Unsteadiness | Falls | Stiff legs | Stiff legs | Stiff legs | Falls | Toe walking, tripping |
| Disease duration (years) | 27 | 48 | 22 | 29 | 2 | 25 | ? | 34 | 24 | 33 | 29 | 5 | 19 | 45 | 12 |
| Disability score (max 7) | 4/7 | 5/7 | 7/7 | 3/7 | NA | 3/7 | 0/7 | 6/7 | 4/7 | 3/7 | 4/7 | 4/7 | 3/7 | 5/7 | 4/7 |
| Spasticity at gait | Severe | Severe | Severe | Moderate | NA | Moderate | None | Severe | Severe | Moderate | Severe | Moderate | Moderate | Moderate | Mild |
| Spasticity at rest | Mild Ashworth 1/4 | Mild | Severe | Mild | NA | Mild | None | None Ashworth 0/4 | Moderate | Moderate Ashworth 2/4 | Mild Ashworth 2/4 | Mild | Mild | Severe | Mild |
| Weakness | Distal > proximal | Diffuse (3/5 MRC grading 4 limbs) | Proximal, distal | Distal > proximal (4 limbs) | L > R | Distal (3/5) > proximal | None | Distal | Distal > proximal | Proximal, distal | Proximal, distal | Proximal, distal | Proximal, distal | Distal > proximal | Distal > proximal |
| Increased reflexes LL | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Yes | Yes | Yes | Yes (ankles decreased) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Normal | Yes | Yes | Yes | Yes | Yes | Yes |
| Extensor plantar response | Indifferent | Yes | Yes | Indifferent | Yes (R) | Yes | R indifferent | Yes | Indifferent | Yes | Yes | Yes | Yes | Yes | Yes |
| Hoffman sign | NA | NA | NA | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | No | NA | NA |
| Decreased vibration sense at ankles | ND | ND | Yes | Mild | NA | No | No | No | Mild | No | No | No | No | Moderate | No |
| Urinary symptoms | Pollakiury | No | Yes | Pollakiury | NA | Yes | No | No | Urinary incontinence | Urinary urgency | Urinary urgency | No | No | Urinary urgency | Urinary urgency |
| Cerebellar gait | No | No | Mild | Mild | NA | Mild | No | Yes (CCFS 0.939) | No | No | No | No | No | No | No |
| Cerebellar signs UL | No | No | Mild | No | NA | No | No | Yes | No | No | No | No | No | No | No |
| Dysarthria | Spastic | No | Anarthria (age 70) | Spastic | NA | Spastic | No | No | Yes | No | No | No | No | No | No |
| Cognitive impairment | No | No | Dementia (age 69) | No | Mild memory impairment | Mild memory impairment | No | No | No | No | No | No | No | No | No |
| Cutaneous findings | No | No | No | No | NA | No | No | No | No | No | No | No | No | Chronic cellulitis changes | No |
| Ocular findings | NA | NA | NA | NA | NA | NA | Congenital cataract | NA | NA | NA | NA | NA | Cataract (age 8 months) | Bilateral senile cataracts (69) | NA |
| Other clinical features | Pes cavus, neuropathic pain | Neuropathic pain | Low back pain, neuropathic pain (LL) | Pes cavus, infantile psychosis, mild horizontal nystagmus | Neuropathic pain (LL), gaze nystagmus, neurosensory deafness, temporal epilepsy | Mild pes cavus | Pes cavus | Pain (knee and back) | Pes cavus | Pes cavus, tremor (left UL) | Mitral leak | Gastrointestinal reflux | Resting tremor, uterine cancer (69) | Pes cavus, hip replacement, groin pain | |
| MRI | Normal brain MRI (2002) | ND | ND | Mild CC atrophy | Normal brain MRI | Large cisterna magna on MRI | NA | White matter anomalies, pons hypersignal, thin dorsal cord | Thin dorsal cord | NA | Left arachnoidien cyst | NA | NA | Cortical atrophy (frontoparietal predominance), chronic microvascular disease (69) | Spinal cord atrophy |
| Other paraclinical tests | ENMG: motor neuronopathy (2009) | NA | NA | ENMG: motor neuronopathy (2014) | Normal ENMG | ENMG: motor neuronopathy (LL, L>R) (2004) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
CC = corpus callosum; CCFS = composite cerebellar functional severity score; ENMG = electro-neuro-myogram; HC = head circumference; ID = intellectual delay; L = left; LL = lower limbs; MD = motor delay; MRC = medical research council; NA = not available; ND = not done; PMD = psychomotor delay; R = right; UL = upper limbs.
In patients from autosomal recessive HSP Families FSP856 and SR45 (Table 3), cognitive impairment was primary, together with moderate to severe early onset HSP. Three of four patients in Family SR45 had a continuum of presentations from tetraparesis to tetraplegia, and two showed pseudobulbar palsy. Both brothers from Family FSP856 had upper limb postural tremor suggesting cerebellar involvement. They also presented microcephaly and facial dysmorphism (2/2), while patients in Family SR45 had short stature (4/4), cataract (1/4) and facial dysmorphism (3/4). Brain MRI showed a thin corpus callosum in patients from Family SR45 but was normal in one patient from Family FSP856 (Supplementary Fig. 2). Of note, both the father and the mother in Family FSP856 carry the heterozygous mutation, while none of them show clinical signs on examination.
Clinical characteristics of patients with ALDH18A1 mutations: autosomal recessive families
| Family N°/Origin | FSP856/Spain | SR45/Portugal | ||||
|---|---|---|---|---|---|---|
| ALDH18A1 variant/Inheritance | p.Asp715His, p.Asp715His/Recessive | p.Arg128His, p.Leu637Pro/Recessive | ||||
| Individual N° (sex) | 18 (M) | 19 (M) | II:3 (F) | II:4 (F) | II:6 (M) | II:9 (F) |
| Age at exam (years) | 42 | 39 | 44 | 49 | 46 | 43 |
| Age at onset (Years) | 7 | 7 | <1 | <1 | <1 | <1 |
| Symptoms at onset | Toe walking, ID, speech delay | ID, gait difficulties | MD, growth retardation | MD, growth retardation | PMD | Severe MD, growth retardation |
| Disease duration (years) | 35 | 32 | 44 | 49 | 46 | 43 |
| Disability score (max 7) | 6/7 | 3/7 | 7/7 | 7/7 | 6/7 | 7/7 |
| Spasticity at gait | Severe | Moderate | Severe, ambulation lost at 30 | Severe | Severe, ambulation lost at 15 | Severe, no ambulation |
| Spasticity at rest | Severe | Moderate | Severe Ashworth 4/4 | Severe Ashworth 3/4 UL; 4/4 LL | Severe Ashworth 4/4 LL; 2/4 UL | Severe Ashworth 4/4 |
| Weakness | Proximal, distal | No | Tetraplegia, drop-feet | Tetraparesia, drop-feet | MRC grading LL 0/5, UL 4/5 | Tetraplegia |
| Increased reflexes LL | Yes | Yes | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Not awakened |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Not awakened |
| Extensor plantar response | Yes | Yes | Yes | Yes | Yes | Indifferent |
| Hoffman sign | NA | NA | NA | NA | NA | NA |
| Decreased vibration sense at ankles | Mild | Mild | NA | NA | No | NA |
| Urinary symptoms | Urinary retention, pollakiuria | No | Incontinence | Incontinence | Incontinence | Incontinence |
| Cerebellar gait | NA | No | No | No | No | NA |
| Cerebellar signs UL | Postural tremor | Postural tremor | No | No | No | NA |
| Dysarthria | No | No | Pseudobulbar, loss of speech (age 30) | Pseudobulbar | Spastic | No speech ever |
| Cognitive impairment | Intellectual deficit IQ 49 | Intellectual deficit IQ 35‐50 | ID, progressive deterioration | ID, progressive deterioration | ID, progressive deterioration | Profound |
| Cutaneous findings | No | No | No | No | No | No |
| Ocular findings | NA | NA | NA | NA | Probable cataract (on neurological examination) | NA |
| Other clinical features | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly, growth retardation, facial dysmorphy; generalized muscle atrophy | Microcephaly, short stature, generalized muscle atrophy | Mild facial dysmorphy, distal amyotrophy (UL, LL) | Severe growth retardation, cyphoscoliosis, archaic reflexes, extreme muscle atrophy |
| MRI | Normal brain MRI | NA | NA | NA | CC atrophy, periventricular white matter anomalies, mild cortical atrophy | NA |
| Family N°/Origin | FSP856/Spain | SR45/Portugal | ||||
|---|---|---|---|---|---|---|
| ALDH18A1 variant/Inheritance | p.Asp715His, p.Asp715His/Recessive | p.Arg128His, p.Leu637Pro/Recessive | ||||
| Individual N° (sex) | 18 (M) | 19 (M) | II:3 (F) | II:4 (F) | II:6 (M) | II:9 (F) |
| Age at exam (years) | 42 | 39 | 44 | 49 | 46 | 43 |
| Age at onset (Years) | 7 | 7 | <1 | <1 | <1 | <1 |
| Symptoms at onset | Toe walking, ID, speech delay | ID, gait difficulties | MD, growth retardation | MD, growth retardation | PMD | Severe MD, growth retardation |
| Disease duration (years) | 35 | 32 | 44 | 49 | 46 | 43 |
| Disability score (max 7) | 6/7 | 3/7 | 7/7 | 7/7 | 6/7 | 7/7 |
| Spasticity at gait | Severe | Moderate | Severe, ambulation lost at 30 | Severe | Severe, ambulation lost at 15 | Severe, no ambulation |
| Spasticity at rest | Severe | Moderate | Severe Ashworth 4/4 | Severe Ashworth 3/4 UL; 4/4 LL | Severe Ashworth 4/4 LL; 2/4 UL | Severe Ashworth 4/4 |
| Weakness | Proximal, distal | No | Tetraplegia, drop-feet | Tetraparesia, drop-feet | MRC grading LL 0/5, UL 4/5 | Tetraplegia |
| Increased reflexes LL | Yes | Yes | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Not awakened |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Not awakened |
| Extensor plantar response | Yes | Yes | Yes | Yes | Yes | Indifferent |
| Hoffman sign | NA | NA | NA | NA | NA | NA |
| Decreased vibration sense at ankles | Mild | Mild | NA | NA | No | NA |
| Urinary symptoms | Urinary retention, pollakiuria | No | Incontinence | Incontinence | Incontinence | Incontinence |
| Cerebellar gait | NA | No | No | No | No | NA |
| Cerebellar signs UL | Postural tremor | Postural tremor | No | No | No | NA |
| Dysarthria | No | No | Pseudobulbar, loss of speech (age 30) | Pseudobulbar | Spastic | No speech ever |
| Cognitive impairment | Intellectual deficit IQ 49 | Intellectual deficit IQ 35‐50 | ID, progressive deterioration | ID, progressive deterioration | ID, progressive deterioration | Profound |
| Cutaneous findings | No | No | No | No | No | No |
| Ocular findings | NA | NA | NA | NA | Probable cataract (on neurological examination) | NA |
| Other clinical features | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly, growth retardation, facial dysmorphy; generalized muscle atrophy | Microcephaly, short stature, generalized muscle atrophy | Mild facial dysmorphy, distal amyotrophy (UL, LL) | Severe growth retardation, cyphoscoliosis, archaic reflexes, extreme muscle atrophy |
| MRI | Normal brain MRI | NA | NA | NA | CC atrophy, periventricular white matter anomalies, mild cortical atrophy | NA |
CC = corpus callosum; CCFS = composite cerebellar functional severity score; ENMG = electro-neuro-myogram; HC = head circumference; ID = intellectual delay; L = left; LL = lower limbs; MD = motor delay; MRC = medical research council; NA = not available; PMD = psychomotor delay; R = right; UL = upper limbs.
Clinical characteristics of patients with ALDH18A1 mutations: autosomal recessive families
| Family N°/Origin | FSP856/Spain | SR45/Portugal | ||||
|---|---|---|---|---|---|---|
| ALDH18A1 variant/Inheritance | p.Asp715His, p.Asp715His/Recessive | p.Arg128His, p.Leu637Pro/Recessive | ||||
| Individual N° (sex) | 18 (M) | 19 (M) | II:3 (F) | II:4 (F) | II:6 (M) | II:9 (F) |
| Age at exam (years) | 42 | 39 | 44 | 49 | 46 | 43 |
| Age at onset (Years) | 7 | 7 | <1 | <1 | <1 | <1 |
| Symptoms at onset | Toe walking, ID, speech delay | ID, gait difficulties | MD, growth retardation | MD, growth retardation | PMD | Severe MD, growth retardation |
| Disease duration (years) | 35 | 32 | 44 | 49 | 46 | 43 |
| Disability score (max 7) | 6/7 | 3/7 | 7/7 | 7/7 | 6/7 | 7/7 |
| Spasticity at gait | Severe | Moderate | Severe, ambulation lost at 30 | Severe | Severe, ambulation lost at 15 | Severe, no ambulation |
| Spasticity at rest | Severe | Moderate | Severe Ashworth 4/4 | Severe Ashworth 3/4 UL; 4/4 LL | Severe Ashworth 4/4 LL; 2/4 UL | Severe Ashworth 4/4 |
| Weakness | Proximal, distal | No | Tetraplegia, drop-feet | Tetraparesia, drop-feet | MRC grading LL 0/5, UL 4/5 | Tetraplegia |
| Increased reflexes LL | Yes | Yes | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Not awakened |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Not awakened |
| Extensor plantar response | Yes | Yes | Yes | Yes | Yes | Indifferent |
| Hoffman sign | NA | NA | NA | NA | NA | NA |
| Decreased vibration sense at ankles | Mild | Mild | NA | NA | No | NA |
| Urinary symptoms | Urinary retention, pollakiuria | No | Incontinence | Incontinence | Incontinence | Incontinence |
| Cerebellar gait | NA | No | No | No | No | NA |
| Cerebellar signs UL | Postural tremor | Postural tremor | No | No | No | NA |
| Dysarthria | No | No | Pseudobulbar, loss of speech (age 30) | Pseudobulbar | Spastic | No speech ever |
| Cognitive impairment | Intellectual deficit IQ 49 | Intellectual deficit IQ 35‐50 | ID, progressive deterioration | ID, progressive deterioration | ID, progressive deterioration | Profound |
| Cutaneous findings | No | No | No | No | No | No |
| Ocular findings | NA | NA | NA | NA | Probable cataract (on neurological examination) | NA |
| Other clinical features | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly, growth retardation, facial dysmorphy; generalized muscle atrophy | Microcephaly, short stature, generalized muscle atrophy | Mild facial dysmorphy, distal amyotrophy (UL, LL) | Severe growth retardation, cyphoscoliosis, archaic reflexes, extreme muscle atrophy |
| MRI | Normal brain MRI | NA | NA | NA | CC atrophy, periventricular white matter anomalies, mild cortical atrophy | NA |
| Family N°/Origin | FSP856/Spain | SR45/Portugal | ||||
|---|---|---|---|---|---|---|
| ALDH18A1 variant/Inheritance | p.Asp715His, p.Asp715His/Recessive | p.Arg128His, p.Leu637Pro/Recessive | ||||
| Individual N° (sex) | 18 (M) | 19 (M) | II:3 (F) | II:4 (F) | II:6 (M) | II:9 (F) |
| Age at exam (years) | 42 | 39 | 44 | 49 | 46 | 43 |
| Age at onset (Years) | 7 | 7 | <1 | <1 | <1 | <1 |
| Symptoms at onset | Toe walking, ID, speech delay | ID, gait difficulties | MD, growth retardation | MD, growth retardation | PMD | Severe MD, growth retardation |
| Disease duration (years) | 35 | 32 | 44 | 49 | 46 | 43 |
| Disability score (max 7) | 6/7 | 3/7 | 7/7 | 7/7 | 6/7 | 7/7 |
| Spasticity at gait | Severe | Moderate | Severe, ambulation lost at 30 | Severe | Severe, ambulation lost at 15 | Severe, no ambulation |
| Spasticity at rest | Severe | Moderate | Severe Ashworth 4/4 | Severe Ashworth 3/4 UL; 4/4 LL | Severe Ashworth 4/4 LL; 2/4 UL | Severe Ashworth 4/4 |
| Weakness | Proximal, distal | No | Tetraplegia, drop-feet | Tetraparesia, drop-feet | MRC grading LL 0/5, UL 4/5 | Tetraplegia |
| Increased reflexes LL | Yes | Yes | Yes (ankles abolished) | Yes (ankles abolished) | Yes (ankles abolished) | Not awakened |
| Increased reflexes UL | Yes | Yes | Yes | Yes | Yes | Not awakened |
| Extensor plantar response | Yes | Yes | Yes | Yes | Yes | Indifferent |
| Hoffman sign | NA | NA | NA | NA | NA | NA |
| Decreased vibration sense at ankles | Mild | Mild | NA | NA | No | NA |
| Urinary symptoms | Urinary retention, pollakiuria | No | Incontinence | Incontinence | Incontinence | Incontinence |
| Cerebellar gait | NA | No | No | No | No | NA |
| Cerebellar signs UL | Postural tremor | Postural tremor | No | No | No | NA |
| Dysarthria | No | No | Pseudobulbar, loss of speech (age 30) | Pseudobulbar | Spastic | No speech ever |
| Cognitive impairment | Intellectual deficit IQ 49 | Intellectual deficit IQ 35‐50 | ID, progressive deterioration | ID, progressive deterioration | ID, progressive deterioration | Profound |
| Cutaneous findings | No | No | No | No | No | No |
| Ocular findings | NA | NA | NA | NA | Probable cataract (on neurological examination) | NA |
| Other clinical features | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly (HC 52 cm, −2 SD), facial dysmorphy | Microcephaly, growth retardation, facial dysmorphy; generalized muscle atrophy | Microcephaly, short stature, generalized muscle atrophy | Mild facial dysmorphy, distal amyotrophy (UL, LL) | Severe growth retardation, cyphoscoliosis, archaic reflexes, extreme muscle atrophy |
| MRI | Normal brain MRI | NA | NA | NA | CC atrophy, periventricular white matter anomalies, mild cortical atrophy | NA |
CC = corpus callosum; CCFS = composite cerebellar functional severity score; ENMG = electro-neuro-myogram; HC = head circumference; ID = intellectual delay; L = left; LL = lower limbs; MD = motor delay; MRC = medical research council; NA = not available; PMD = psychomotor delay; R = right; UL = upper limbs.
Biochemical analyses in blood and dermal fibroblasts
Blood amino acid chromatography (Table 4) was performed in four patients from two unrelated autosomal dominant families. Three family members of Family FSP410 had low citrulline levels, associated with low proline (2/3), ornithine (1/3, 1/3 borderline), and borderline levels of arginine (3/3). A profound decrease of plasma citrulline was also observed in Patient FSP429‐21, associated with low arginine and borderline ornithine levels. By contrast, amino acid levels were normal in Patient FSP856‐18 with autosomal recessive inheritance (data not shown). In all tested patients, levels of other amino acids in blood and all amino acids in urine were within normal range. The amino acids profiles from the patients in this study, along with the two first patients reported with ALDH18A1 mutations (Baumgartner et al., 2005), were compared to 5023 profiles from Necker Hospital full cohort of patients aged over 15 years that had no known metabolic disorders. The comparison showed that low levels of ornithine, citrulline, arginine and proline (Fig. 3A) were significantly predictive of ALDH18A1 mutations (area under the receiver operating characteristic curve 97%, with 95% confidence interval: 96–99%, Supplementary Fig. 3A). Adding elevated glutamine as a covariate may increase prediction (Supplementary Fig. 3B).
Amino acid levels on plasma chromatography
| Patient | Age | Proline | Ornithine | Citrulline | Arginine | Valine | Leucine | Isoleucine | Threonine | Glutamine | Alanine | Methionine | Lysine |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FSP410‐032 | 43 | 163 | 35 | 12 | 58 | 190 | 116 | 51 | 69 | 517 | 237 | 22 | 146 |
| FSP410‐029 | 45 | 117 | 83 | 8 | 63 | 215 | 110 | 61 | 208 | 740 | 400 | 23 | 201 |
| FSP410‐013 | 70 | 73 | 46 | 8 | 54 | 178 | 108 | 51 | 91 | 632 | 277 | 18 | 178 |
| FSP429‐021 | 40 | 209 | 54 | 1 | 28 | 267 | 119 | 52 | 114 | 470 | 304 | 19 | 175 |
| Normal ranges | 150–220 | 50–100 | 20–35 | 60–100 | 210–280 | 100–150 | 50–80 | 95–195 | 430–670 | 285–415 | 20–35 | 155–230 |
| Patient | Age | Proline | Ornithine | Citrulline | Arginine | Valine | Leucine | Isoleucine | Threonine | Glutamine | Alanine | Methionine | Lysine |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FSP410‐032 | 43 | 163 | 35 | 12 | 58 | 190 | 116 | 51 | 69 | 517 | 237 | 22 | 146 |
| FSP410‐029 | 45 | 117 | 83 | 8 | 63 | 215 | 110 | 61 | 208 | 740 | 400 | 23 | 201 |
| FSP410‐013 | 70 | 73 | 46 | 8 | 54 | 178 | 108 | 51 | 91 | 632 | 277 | 18 | 178 |
| FSP429‐021 | 40 | 209 | 54 | 1 | 28 | 267 | 119 | 52 | 114 | 470 | 304 | 19 | 175 |
| Normal ranges | 150–220 | 50–100 | 20–35 | 60–100 | 210–280 | 100–150 | 50–80 | 95–195 | 430–670 | 285–415 | 20–35 | 155–230 |
Values are given in μmol/l. Bold indicates low values, italics indicates borderline values.
Amino acid levels on plasma chromatography
| Patient | Age | Proline | Ornithine | Citrulline | Arginine | Valine | Leucine | Isoleucine | Threonine | Glutamine | Alanine | Methionine | Lysine |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FSP410‐032 | 43 | 163 | 35 | 12 | 58 | 190 | 116 | 51 | 69 | 517 | 237 | 22 | 146 |
| FSP410‐029 | 45 | 117 | 83 | 8 | 63 | 215 | 110 | 61 | 208 | 740 | 400 | 23 | 201 |
| FSP410‐013 | 70 | 73 | 46 | 8 | 54 | 178 | 108 | 51 | 91 | 632 | 277 | 18 | 178 |
| FSP429‐021 | 40 | 209 | 54 | 1 | 28 | 267 | 119 | 52 | 114 | 470 | 304 | 19 | 175 |
| Normal ranges | 150–220 | 50–100 | 20–35 | 60–100 | 210–280 | 100–150 | 50–80 | 95–195 | 430–670 | 285–415 | 20–35 | 155–230 |
| Patient | Age | Proline | Ornithine | Citrulline | Arginine | Valine | Leucine | Isoleucine | Threonine | Glutamine | Alanine | Methionine | Lysine |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FSP410‐032 | 43 | 163 | 35 | 12 | 58 | 190 | 116 | 51 | 69 | 517 | 237 | 22 | 146 |
| FSP410‐029 | 45 | 117 | 83 | 8 | 63 | 215 | 110 | 61 | 208 | 740 | 400 | 23 | 201 |
| FSP410‐013 | 70 | 73 | 46 | 8 | 54 | 178 | 108 | 51 | 91 | 632 | 277 | 18 | 178 |
| FSP429‐021 | 40 | 209 | 54 | 1 | 28 | 267 | 119 | 52 | 114 | 470 | 304 | 19 | 175 |
| Normal ranges | 150–220 | 50–100 | 20–35 | 60–100 | 210–280 | 100–150 | 50–80 | 95–195 | 430–670 | 285–415 | 20–35 | 155–230 |
Values are given in μmol/l. Bold indicates low values, italics indicates borderline values.
In cultured dermal fibroblasts from two patients of Family FSP410, proline biosynthesis was estimated by the stable isotope label enrichment in proline after loading for 13C5-labelled glutamine (Baumgartner et al., 2005). Assuming a stationary phase at 18-h incubation, the two patients harbouring ALDH18A1 mutations had 42% residual flux (stable isotope ratios 0.68 + 0.08 and 0.95 + 0.06, average: 0.81) compared to age-matched healthy controls (stable isotope ratio 1.83 ± 0.05 and 2.01 ± 0.14, average: 1.92) (Fig. 3B). This confirms a reduction of proline biosynthesis consistent with an enzymatic deficiency of P5CS.
Discussion
Here we report seven pedigrees with ALDH18A1 mutations. Two families with biallelic mutations presented complex autosomal recessive HSP with marked cognitive impairment and no cutaneous involvement. Remarkably, we also identified, in three independent families, monoallelic ALDH18A1 mutations segregating with autosomal dominant HSP, together with low plasma levels of specific amino acids, especially citrulline, and ex vivo demonstration of a metabolic block at the level of P5CS. Two additional sporadic cases harboured monoallelic mutations in ALDH18A1. Therefore, this study broadens the clinical spectrum of autosomal recessive ALDH18A1-linked pathology, and describes a new mode of inheritance associated with a potential trait biomarker in plasma. Expanding phenotype and describing new modes of transmission becomes more and more frequent in HSP (Esteves et al., 2014; Tesson et al., 2015); such bimodal transmission has also been described long ago, for example in human myotonia, caused by either dominant or recessive mutations of CLCN1 (Koch et al., 1992).
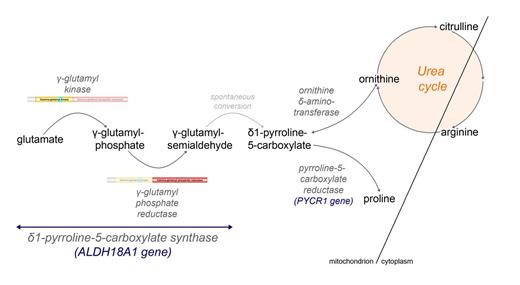
ALDH18A1 encodes P5CS, a bifunctional enzyme that catalyses the first common step in proline and ornithine biosynthesis (Hu et al., 1999, 2008b). This enzyme, located in the mitochondrial inner membrane, catalyses the conversion of glutamate to gamma-glutamyl semi-aldehyde (Fig. 3), which spontaneously converts to pyrroline-5-carboxylate (P5C). P5C can thereafter be metabolized to proline by P5C reductase (PYCR1), or to ornithine, citrulline and finally arginine through the urea cycle (Fig. 3). P5CS comprises two domains, with different enzymatic activities: an N-terminal ATP-dependant gamma-glutamyl kinase domain, responsible for the glutamate phosphorylation to gamma-glutamyl phosphate, and a C-terminal NADPH-dependant gamma-glutamyl phosphate reductase domain, which catalyses the reduction and conversion to gamma-glutamyl semi-aldehyde (Fichman et al., 2014). Although P5CS expression in the brain is not strong (Hu et al., 1999), it has a measurable activity (Hu et al., 2008b).
ALDH18A1 mutations account for a phenotypic spectrum ranging from pure autosomal dominant to complex autosomal recessive HSP
In humans, mutations in two genes coding for enzymes involved in proline biosynthesis have been described in overlapping neurocutaneous syndromes; ALDH18A1 and PYCR1. PYCR1 mutations are implicated in an autosomal recessive neurocutaneous syndrome with mental retardation, cutis laxa, joint hyperlaxity, progeroid dysmorphy, growth retardation, microcephaly, and corpus callosum dysgenesis (Reversade et al., 2009; Dimopoulou et al., 2013; Gardeitchik et al., 2014). These patients present normal amino acid levels (Reversade et al., 2009).
As for ALDH18A1, the first two patients with demonstrated P5CS deficiency carried a homozygous missense mutation in the glutamate-5-kinase domain of the protein (Baumgartner et al., 2000, 2005), and presented a syndrome associating cutis laxa, joint hyperlaxity, hypotonia, developmental delay, cataract, pyramidal signs and plasma amino acids anomalies—low proline, ornithine, citrulline and arginine. Subsequently, Bicknell et al. (2008) reported four siblings with a homozygous missense mutation in the gamma-glutamyl phosphate reductase domain of P5CS; the clinical picture was very similar with the exception of plasma amino acids levels, which showed no abnormalities. Until now, 15 patients with ALDH18A1 biallelic mutations have been reported (Baumgartner et al., 2000, 2005; Bicknell et al., 2008; Skidmore et al., 2011; Martinelli et al., 2012; Zampatti et al., 2012; Fischer et al., 2014; Gardeitchik et al., 2014; Handley et al., 2014; Wolthuis et al., 2014). Developmental delay, failure to thrive, hypotonia, cognitive impairment, cutis laxa, joint hyperlaxity, growth retardation and cataract are cardinal signs (Table 5). Dystonia, seizures in infancy, thin corpus callosum, pes planus, hip dislocation, bone malformations, gastro-oesophageal reflux and inguinal hernia are frequent signs orientating towards the molecular diagnosis. Pyramidal signs are often reported (12/15), but not cardinal; most of the patients presented brisk reflexes and pyramidal syndrome was judged severe in three only.
Summary of clinical characteristics of published families with ALDH18A1 mutations
| Reference | Baumgartner et al., 2000, 2005 | Bicknell et al., 2008 | Skidmore et al., 2011 | Martinelli et al., 2012 | Zampatti et al., 2012; Gardeitchik et al., 2014 | Wolthuis et al., 2014 | Fischer et al., 2014 | Fischer et al., 2014 | Handley et al., 2014 | This study (FSP856) | This study (SR45) | This study (FSP410) | This study (FSP429) | This study (FSP470) | This study (GSHSP44) | This study (25014) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutations (protein level) | R84Q/R84Q | H784Y/H784Y | c.1923+1 G>A/c.1923+1 G>A | G93R/T299I | S742I/R425C | Y782C/Y782C | V601Gfs*/V601Gfs* | L711Cfs*/L711Cfs* | R749Q/R765Q | D715H/D715H | R128H/L637P | V120A | R665L | R252Q | S652F | R252Q |
| Functional effect | Reduced P5CS activity | Normal PRO and ORN synthesis | Reduced fibroblasts proliferation and P5CS expression | Decreased P5CS activity, dissociation to dimers or monomers; no mitochondrial alterations in cultured fibroblasts | Abolished P5CS expression, swollen mitochondria in fibroblasts (electron microscopy) | Reduced P5CS expression, lipid droplet enlargement | NA | NA | Lowered PRO synthesis in fibroblasts | NA | NA | NA | NA | |||
| Low plasma amino acids levels (other findings) | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal (mild post-prandial hypoornithinaemia in 2/3) | Normal | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal | Normal | CITRU, ARG | NA | Normal | NA | NA | CITRU (3/3), PRO (2/3), low or borderline ORN(2/3), and ARG (3/3) | CITRU, ARG, (borderline ORN) | NA | NA | NA |
| Neurological signs at onset | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | Intellectual deficiency | Global delay, growth retardation | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | |
| Pyramidal signs | Yes (2/2), severe in 1 | Hypertonicity LL (4/4), UL (2/4) | Yes | NA | Yes | Brisk reflexes | NA | NA | Brisk reflexes | Moderate to severe | Severe, tetraplegia (2/4) or tetraparesis (1/4) | Moderate to severe | Severe | Moderate to severe | Mild | Moderate to severe |
| Other neurological findings | Peripheral axonal neuropathy, seizures in infancy (1/2), distal dystonia (1/2) | Seizures in infancy (2/4), absent speech | Flexion contractures of elbows, wrists (32w gestation) | Seizures in infancy | Seizures, dystonia, absent speech | Disseminated tremors, finger contractures | Seizures | Joint contractures | No | No | Mild cerebellar signs (2/6), motor neuropathy (2/6), dementia (1/6) | Cerebellar signs (1/2) | No | No | No | |
| Cutis laxa (other cutaneous findings) | Yes | Yes | Yes (sparse hair) | Yes | Yes (sparse hair) | Yes | Yes | Yes | Yes | No | No | No | No | No | No | Chronic cellulitis changes |
| Skeletal findings | Joint hyperlaxity, pes planus | Joint hyperlaxity, hip dislocation (3/4), kyphoscoliosis | NA | Joint hyperlaxity, pes planus, coxa valga | Joint hypermobility, osteopenia, rib anomalies | Joint laxity, bilateral hip dislocation and dysplasia | Unilateral hip dislocation, kyphosis | Joint hypermobility, congenital luxation of left hip | Bilateral coxa valga | Pes cavus (3/6) | Pes cavus (1/2) | Pes cavus (2/4) | Pes cavus, hip replacement | |||
| Facial dysmorphy | Yes | NA | Yes | Yes | Yes | NA | Yes | NA | Yes | Yes | Yes | No | No | No | No | No |
| Growth retardation/microcephaly | Yes/yes | Yes/yes | Yes/yes | Yes/macrocephaly | Yes/no | Yes/yes | Yes/yes | Yes/yes | Yes/yes | No/yes | Yes/yes | No/no | No/no | No/no | No/no | No |
| Cataracts (other ocular findings) | Yes | Yes 1/4 | (Corneal clouding) | Yes | Yes | (Retinitis pigmentosa) | Yes | (Corneal clouding) | Yes 1/2 | NA | Yes 1/4 | Yes 1/6 | NA | Yes 1/4 | NA | No |
| Gastro-oesophagal reflux | 2/2 | NA | NA | NA | Yes | NA | NA | NA | NA | NA | NA | NA | NA | 1/4 | NA | NA |
| Inguinal hernia | NA | 1/4 | Bilateral | Bilateral | 1/2 | Yes | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Vascular findings | NA | NA | Arterial tortuosities | NA | NA | NA | Cardiovascular malformations | Narrow aortic arch, probable aneurysm | NA | NA | NA | NA | NA | Mitral leak (1/4) | NA | NA |
| Other | Abnormal fat distribution, undescended testis | Bilateral hearing loss | Undescended testis | Infantile psychosis (1/6) | ||||||||||||
| MRI | NA | Paucity of WM, thin CC | NA | Mild cortical atrophy, thin CC, tortuosity of brain vessels | Cerebellar vermis hypoplasia, thin CC | Thin CC | Normal | Agenesis of CC | Thin CC, prominent lateral and 3rd ventricles, cerebellar hypoplasia, mildly delayed myelination | Normal | Thin CC, mild cerebellar atrophy, WM anomalies | Mild CC atrophy (1/3) | WM anomalies, thin dorsal cord, pons hypersignal (1/2) | Left arachnoidien cyst (1/4) | Spinal cord atrophy | Cortical atrophy (frontoparietal predominance) |
| Reference | Baumgartner et al., 2000, 2005 | Bicknell et al., 2008 | Skidmore et al., 2011 | Martinelli et al., 2012 | Zampatti et al., 2012; Gardeitchik et al., 2014 | Wolthuis et al., 2014 | Fischer et al., 2014 | Fischer et al., 2014 | Handley et al., 2014 | This study (FSP856) | This study (SR45) | This study (FSP410) | This study (FSP429) | This study (FSP470) | This study (GSHSP44) | This study (25014) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutations (protein level) | R84Q/R84Q | H784Y/H784Y | c.1923+1 G>A/c.1923+1 G>A | G93R/T299I | S742I/R425C | Y782C/Y782C | V601Gfs*/V601Gfs* | L711Cfs*/L711Cfs* | R749Q/R765Q | D715H/D715H | R128H/L637P | V120A | R665L | R252Q | S652F | R252Q |
| Functional effect | Reduced P5CS activity | Normal PRO and ORN synthesis | Reduced fibroblasts proliferation and P5CS expression | Decreased P5CS activity, dissociation to dimers or monomers; no mitochondrial alterations in cultured fibroblasts | Abolished P5CS expression, swollen mitochondria in fibroblasts (electron microscopy) | Reduced P5CS expression, lipid droplet enlargement | NA | NA | Lowered PRO synthesis in fibroblasts | NA | NA | NA | NA | |||
| Low plasma amino acids levels (other findings) | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal (mild post-prandial hypoornithinaemia in 2/3) | Normal | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal | Normal | CITRU, ARG | NA | Normal | NA | NA | CITRU (3/3), PRO (2/3), low or borderline ORN(2/3), and ARG (3/3) | CITRU, ARG, (borderline ORN) | NA | NA | NA |
| Neurological signs at onset | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | Intellectual deficiency | Global delay, growth retardation | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | |
| Pyramidal signs | Yes (2/2), severe in 1 | Hypertonicity LL (4/4), UL (2/4) | Yes | NA | Yes | Brisk reflexes | NA | NA | Brisk reflexes | Moderate to severe | Severe, tetraplegia (2/4) or tetraparesis (1/4) | Moderate to severe | Severe | Moderate to severe | Mild | Moderate to severe |
| Other neurological findings | Peripheral axonal neuropathy, seizures in infancy (1/2), distal dystonia (1/2) | Seizures in infancy (2/4), absent speech | Flexion contractures of elbows, wrists (32w gestation) | Seizures in infancy | Seizures, dystonia, absent speech | Disseminated tremors, finger contractures | Seizures | Joint contractures | No | No | Mild cerebellar signs (2/6), motor neuropathy (2/6), dementia (1/6) | Cerebellar signs (1/2) | No | No | No | |
| Cutis laxa (other cutaneous findings) | Yes | Yes | Yes (sparse hair) | Yes | Yes (sparse hair) | Yes | Yes | Yes | Yes | No | No | No | No | No | No | Chronic cellulitis changes |
| Skeletal findings | Joint hyperlaxity, pes planus | Joint hyperlaxity, hip dislocation (3/4), kyphoscoliosis | NA | Joint hyperlaxity, pes planus, coxa valga | Joint hypermobility, osteopenia, rib anomalies | Joint laxity, bilateral hip dislocation and dysplasia | Unilateral hip dislocation, kyphosis | Joint hypermobility, congenital luxation of left hip | Bilateral coxa valga | Pes cavus (3/6) | Pes cavus (1/2) | Pes cavus (2/4) | Pes cavus, hip replacement | |||
| Facial dysmorphy | Yes | NA | Yes | Yes | Yes | NA | Yes | NA | Yes | Yes | Yes | No | No | No | No | No |
| Growth retardation/microcephaly | Yes/yes | Yes/yes | Yes/yes | Yes/macrocephaly | Yes/no | Yes/yes | Yes/yes | Yes/yes | Yes/yes | No/yes | Yes/yes | No/no | No/no | No/no | No/no | No |
| Cataracts (other ocular findings) | Yes | Yes 1/4 | (Corneal clouding) | Yes | Yes | (Retinitis pigmentosa) | Yes | (Corneal clouding) | Yes 1/2 | NA | Yes 1/4 | Yes 1/6 | NA | Yes 1/4 | NA | No |
| Gastro-oesophagal reflux | 2/2 | NA | NA | NA | Yes | NA | NA | NA | NA | NA | NA | NA | NA | 1/4 | NA | NA |
| Inguinal hernia | NA | 1/4 | Bilateral | Bilateral | 1/2 | Yes | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Vascular findings | NA | NA | Arterial tortuosities | NA | NA | NA | Cardiovascular malformations | Narrow aortic arch, probable aneurysm | NA | NA | NA | NA | NA | Mitral leak (1/4) | NA | NA |
| Other | Abnormal fat distribution, undescended testis | Bilateral hearing loss | Undescended testis | Infantile psychosis (1/6) | ||||||||||||
| MRI | NA | Paucity of WM, thin CC | NA | Mild cortical atrophy, thin CC, tortuosity of brain vessels | Cerebellar vermis hypoplasia, thin CC | Thin CC | Normal | Agenesis of CC | Thin CC, prominent lateral and 3rd ventricles, cerebellar hypoplasia, mildly delayed myelination | Normal | Thin CC, mild cerebellar atrophy, WM anomalies | Mild CC atrophy (1/3) | WM anomalies, thin dorsal cord, pons hypersignal (1/2) | Left arachnoidien cyst (1/4) | Spinal cord atrophy | Cortical atrophy (frontoparietal predominance) |
ARG = arginine; CC = corpus callosum; CITRU = citrulline; DD = developmental delay; LL = lower limbs; NA = not available; ORN = ornithine; PRO = proline; UL = upper limbs; WM = white matter.
Summary of clinical characteristics of published families with ALDH18A1 mutations
| Reference | Baumgartner et al., 2000, 2005 | Bicknell et al., 2008 | Skidmore et al., 2011 | Martinelli et al., 2012 | Zampatti et al., 2012; Gardeitchik et al., 2014 | Wolthuis et al., 2014 | Fischer et al., 2014 | Fischer et al., 2014 | Handley et al., 2014 | This study (FSP856) | This study (SR45) | This study (FSP410) | This study (FSP429) | This study (FSP470) | This study (GSHSP44) | This study (25014) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutations (protein level) | R84Q/R84Q | H784Y/H784Y | c.1923+1 G>A/c.1923+1 G>A | G93R/T299I | S742I/R425C | Y782C/Y782C | V601Gfs*/V601Gfs* | L711Cfs*/L711Cfs* | R749Q/R765Q | D715H/D715H | R128H/L637P | V120A | R665L | R252Q | S652F | R252Q |
| Functional effect | Reduced P5CS activity | Normal PRO and ORN synthesis | Reduced fibroblasts proliferation and P5CS expression | Decreased P5CS activity, dissociation to dimers or monomers; no mitochondrial alterations in cultured fibroblasts | Abolished P5CS expression, swollen mitochondria in fibroblasts (electron microscopy) | Reduced P5CS expression, lipid droplet enlargement | NA | NA | Lowered PRO synthesis in fibroblasts | NA | NA | NA | NA | |||
| Low plasma amino acids levels (other findings) | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal (mild post-prandial hypoornithinaemia in 2/3) | Normal | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal | Normal | CITRU, ARG | NA | Normal | NA | NA | CITRU (3/3), PRO (2/3), low or borderline ORN(2/3), and ARG (3/3) | CITRU, ARG, (borderline ORN) | NA | NA | NA |
| Neurological signs at onset | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | Intellectual deficiency | Global delay, growth retardation | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | |
| Pyramidal signs | Yes (2/2), severe in 1 | Hypertonicity LL (4/4), UL (2/4) | Yes | NA | Yes | Brisk reflexes | NA | NA | Brisk reflexes | Moderate to severe | Severe, tetraplegia (2/4) or tetraparesis (1/4) | Moderate to severe | Severe | Moderate to severe | Mild | Moderate to severe |
| Other neurological findings | Peripheral axonal neuropathy, seizures in infancy (1/2), distal dystonia (1/2) | Seizures in infancy (2/4), absent speech | Flexion contractures of elbows, wrists (32w gestation) | Seizures in infancy | Seizures, dystonia, absent speech | Disseminated tremors, finger contractures | Seizures | Joint contractures | No | No | Mild cerebellar signs (2/6), motor neuropathy (2/6), dementia (1/6) | Cerebellar signs (1/2) | No | No | No | |
| Cutis laxa (other cutaneous findings) | Yes | Yes | Yes (sparse hair) | Yes | Yes (sparse hair) | Yes | Yes | Yes | Yes | No | No | No | No | No | No | Chronic cellulitis changes |
| Skeletal findings | Joint hyperlaxity, pes planus | Joint hyperlaxity, hip dislocation (3/4), kyphoscoliosis | NA | Joint hyperlaxity, pes planus, coxa valga | Joint hypermobility, osteopenia, rib anomalies | Joint laxity, bilateral hip dislocation and dysplasia | Unilateral hip dislocation, kyphosis | Joint hypermobility, congenital luxation of left hip | Bilateral coxa valga | Pes cavus (3/6) | Pes cavus (1/2) | Pes cavus (2/4) | Pes cavus, hip replacement | |||
| Facial dysmorphy | Yes | NA | Yes | Yes | Yes | NA | Yes | NA | Yes | Yes | Yes | No | No | No | No | No |
| Growth retardation/microcephaly | Yes/yes | Yes/yes | Yes/yes | Yes/macrocephaly | Yes/no | Yes/yes | Yes/yes | Yes/yes | Yes/yes | No/yes | Yes/yes | No/no | No/no | No/no | No/no | No |
| Cataracts (other ocular findings) | Yes | Yes 1/4 | (Corneal clouding) | Yes | Yes | (Retinitis pigmentosa) | Yes | (Corneal clouding) | Yes 1/2 | NA | Yes 1/4 | Yes 1/6 | NA | Yes 1/4 | NA | No |
| Gastro-oesophagal reflux | 2/2 | NA | NA | NA | Yes | NA | NA | NA | NA | NA | NA | NA | NA | 1/4 | NA | NA |
| Inguinal hernia | NA | 1/4 | Bilateral | Bilateral | 1/2 | Yes | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Vascular findings | NA | NA | Arterial tortuosities | NA | NA | NA | Cardiovascular malformations | Narrow aortic arch, probable aneurysm | NA | NA | NA | NA | NA | Mitral leak (1/4) | NA | NA |
| Other | Abnormal fat distribution, undescended testis | Bilateral hearing loss | Undescended testis | Infantile psychosis (1/6) | ||||||||||||
| MRI | NA | Paucity of WM, thin CC | NA | Mild cortical atrophy, thin CC, tortuosity of brain vessels | Cerebellar vermis hypoplasia, thin CC | Thin CC | Normal | Agenesis of CC | Thin CC, prominent lateral and 3rd ventricles, cerebellar hypoplasia, mildly delayed myelination | Normal | Thin CC, mild cerebellar atrophy, WM anomalies | Mild CC atrophy (1/3) | WM anomalies, thin dorsal cord, pons hypersignal (1/2) | Left arachnoidien cyst (1/4) | Spinal cord atrophy | Cortical atrophy (frontoparietal predominance) |
| Reference | Baumgartner et al., 2000, 2005 | Bicknell et al., 2008 | Skidmore et al., 2011 | Martinelli et al., 2012 | Zampatti et al., 2012; Gardeitchik et al., 2014 | Wolthuis et al., 2014 | Fischer et al., 2014 | Fischer et al., 2014 | Handley et al., 2014 | This study (FSP856) | This study (SR45) | This study (FSP410) | This study (FSP429) | This study (FSP470) | This study (GSHSP44) | This study (25014) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutations (protein level) | R84Q/R84Q | H784Y/H784Y | c.1923+1 G>A/c.1923+1 G>A | G93R/T299I | S742I/R425C | Y782C/Y782C | V601Gfs*/V601Gfs* | L711Cfs*/L711Cfs* | R749Q/R765Q | D715H/D715H | R128H/L637P | V120A | R665L | R252Q | S652F | R252Q |
| Functional effect | Reduced P5CS activity | Normal PRO and ORN synthesis | Reduced fibroblasts proliferation and P5CS expression | Decreased P5CS activity, dissociation to dimers or monomers; no mitochondrial alterations in cultured fibroblasts | Abolished P5CS expression, swollen mitochondria in fibroblasts (electron microscopy) | Reduced P5CS expression, lipid droplet enlargement | NA | NA | Lowered PRO synthesis in fibroblasts | NA | NA | NA | NA | |||
| Low plasma amino acids levels (other findings) | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal (mild post-prandial hypoornithinaemia in 2/3) | Normal | ORN, CITRU, ARG, PRO (paradoxal hyperammonaemia) | Normal | Normal | CITRU, ARG | NA | Normal | NA | NA | CITRU (3/3), PRO (2/3), low or borderline ORN(2/3), and ARG (3/3) | CITRU, ARG, (borderline ORN) | NA | NA | NA |
| Neurological signs at onset | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | DD, hypotonia | Intellectual deficiency | Global delay, growth retardation | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | Pyramidal signs | |
| Pyramidal signs | Yes (2/2), severe in 1 | Hypertonicity LL (4/4), UL (2/4) | Yes | NA | Yes | Brisk reflexes | NA | NA | Brisk reflexes | Moderate to severe | Severe, tetraplegia (2/4) or tetraparesis (1/4) | Moderate to severe | Severe | Moderate to severe | Mild | Moderate to severe |
| Other neurological findings | Peripheral axonal neuropathy, seizures in infancy (1/2), distal dystonia (1/2) | Seizures in infancy (2/4), absent speech | Flexion contractures of elbows, wrists (32w gestation) | Seizures in infancy | Seizures, dystonia, absent speech | Disseminated tremors, finger contractures | Seizures | Joint contractures | No | No | Mild cerebellar signs (2/6), motor neuropathy (2/6), dementia (1/6) | Cerebellar signs (1/2) | No | No | No | |
| Cutis laxa (other cutaneous findings) | Yes | Yes | Yes (sparse hair) | Yes | Yes (sparse hair) | Yes | Yes | Yes | Yes | No | No | No | No | No | No | Chronic cellulitis changes |
| Skeletal findings | Joint hyperlaxity, pes planus | Joint hyperlaxity, hip dislocation (3/4), kyphoscoliosis | NA | Joint hyperlaxity, pes planus, coxa valga | Joint hypermobility, osteopenia, rib anomalies | Joint laxity, bilateral hip dislocation and dysplasia | Unilateral hip dislocation, kyphosis | Joint hypermobility, congenital luxation of left hip | Bilateral coxa valga | Pes cavus (3/6) | Pes cavus (1/2) | Pes cavus (2/4) | Pes cavus, hip replacement | |||
| Facial dysmorphy | Yes | NA | Yes | Yes | Yes | NA | Yes | NA | Yes | Yes | Yes | No | No | No | No | No |
| Growth retardation/microcephaly | Yes/yes | Yes/yes | Yes/yes | Yes/macrocephaly | Yes/no | Yes/yes | Yes/yes | Yes/yes | Yes/yes | No/yes | Yes/yes | No/no | No/no | No/no | No/no | No |
| Cataracts (other ocular findings) | Yes | Yes 1/4 | (Corneal clouding) | Yes | Yes | (Retinitis pigmentosa) | Yes | (Corneal clouding) | Yes 1/2 | NA | Yes 1/4 | Yes 1/6 | NA | Yes 1/4 | NA | No |
| Gastro-oesophagal reflux | 2/2 | NA | NA | NA | Yes | NA | NA | NA | NA | NA | NA | NA | NA | 1/4 | NA | NA |
| Inguinal hernia | NA | 1/4 | Bilateral | Bilateral | 1/2 | Yes | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Vascular findings | NA | NA | Arterial tortuosities | NA | NA | NA | Cardiovascular malformations | Narrow aortic arch, probable aneurysm | NA | NA | NA | NA | NA | Mitral leak (1/4) | NA | NA |
| Other | Abnormal fat distribution, undescended testis | Bilateral hearing loss | Undescended testis | Infantile psychosis (1/6) | ||||||||||||
| MRI | NA | Paucity of WM, thin CC | NA | Mild cortical atrophy, thin CC, tortuosity of brain vessels | Cerebellar vermis hypoplasia, thin CC | Thin CC | Normal | Agenesis of CC | Thin CC, prominent lateral and 3rd ventricles, cerebellar hypoplasia, mildly delayed myelination | Normal | Thin CC, mild cerebellar atrophy, WM anomalies | Mild CC atrophy (1/3) | WM anomalies, thin dorsal cord, pons hypersignal (1/2) | Left arachnoidien cyst (1/4) | Spinal cord atrophy | Cortical atrophy (frontoparietal predominance) |
ARG = arginine; CC = corpus callosum; CITRU = citrulline; DD = developmental delay; LL = lower limbs; NA = not available; ORN = ornithine; PRO = proline; UL = upper limbs; WM = white matter.
The patients that we report with biallelic ALDH18A1 mutations (Families FSP856 and SR45) share common features with those previously reported suffering from neurocutaneous syndromes: developmental delay (4/6), intellectual deficiency (6/6), short stature (4/6), facial dysmorphism (6/6), cataract (1/6) and thin corpus callosum (1/3). Of interest is the severity of pyramidal signs, with moderate to severe spasticity (6/6), to tetraplegia (1/6) and pseudobulbar palsy (2/6). Although severe HSP was previously reported in ALDH18A1 patients, its frequency was low (3/15). Moreover, though cutis laxa has been reported to improve or disappear with age in some patients (Bicknell et al., 2008), it was never noted in any of our patients, while it was previously considered as an obligate sign. We therefore are expanding the clinical spectrum of biallelic ALDH18A1 mutations, with emphasis on the pyramidal component alteration; and are establishing that the cutaneous abnormalities may be lacking.
More importantly, we also report the first three families with autosomal dominant HSP segregating ALDH18A1 mutations. The following arguments are in favour of the pathogenicity of the variants we report: (i) the absence of reported mutations in all examined public databases (∼63 000 exomes); (ii) amino acid conservation through phylogenesis; (iii) in silico pathogenicity predictions (Table 1); (iv) location within a putatively linked locus in Family FSP410 (Supplementary Fig. 1); (v) absence of pathogenic variants in all known HSP genes; (vi) multiplicity of the mutated families and multiplicity of patients with the mutations amongst the families; (vii) location within the aspartokinase active site for p.R252Q; (viii) a second occurrence of the p.R252Q mutation with a different haplotype; (ix) abnormal plasma amino acids levels in carriers from two unrelated families (Families FSP410 and FSP429); and (x) reduced glutamate-to-proline metabolic flux in dermal fibroblasts of two patients of Family FSP410 (42% of levels measured in control fibroblasts). However, the clinical picture of these patients largely differs from biallelic mutation carriers. Spasticity is the cardinal and first presentation, and is often pure with few or no accompanying signs. Seven of 15 patients had pes cavus that could be a consequence of the pyramidal syndrome, while pes planus was classically described in autosomal recessive pathology. Interestingly, Patients FSP410‐22 and FSP470‐22 presented with congenital cataract, which is a distinctive sign, when present, in patients with biallelic ALDH18A1 mutations (Baumgartner et al., 2000, 2005; Zampatti et al., 2012). A peripheral neuropathy was demonstrated in three of four patients that had electromyography in Family FSP410, but was clinically present in 9 of 21 patients, as in the first reported patients with biallelic ALDH18A1 mutations (Baumgartner et al., 2005). Cerebellar signs were noted in 4 of 14 autosomal dominant patients, as in two of six patients with biallelic mutations. It has never been reported before, but cerebellar atrophy was evidenced in several reports (Zampatti et al., 2012; Handley et al., 2014). These elements are again in favour of the implication of ALDH18A1 mutations in these cases. Of note, the clinical presentation was not homogeneous among the respective families, especially for Family FSP410. Age, disease duration, lifestyle, and dietary factors are all potential modifiers underlying this variability.
What is the pathogenic mechanism linked to ALDH18A1 mutations?
In autosomal recessive families, all previously reported patients had either homozygous variants or compound heterozygous mutations affecting the same functional domain and, therefore, a presumably complete enzymatic block of one of the P5CS-catalysed reactions (Fig. 3). In addition, abnormal metabolic profiles were only reported in patients with mutations affecting the glutamate-5-kinase domain (Baumgartner et al., 2000; Martinelli et al., 2012) or with a complete abolition of protein expression (Fischer et al., 2014). Consistently, the amino acid profile is normal in Patient FSP856‐18 that harbours mutations that do not affect the glutamate-5-kinase domain. Of note, Family SR45 is the first reported with compound heterozygous missense mutations involving both domains of P5CS; here, a complete enzymatic block is not expected.
In autosomal dominant families, the most striking anomalies are the low to very low citrulline levels in the plasma of all patients tested, regardless of the protein domain affected by the mutation, together with a less consistent, decreased ornithine, arginine and proline levels. The sum of age-normalized values of these four amino acids is obviously reduced in our patients, as well as in the first family in which the disorder was identified (Fig. 2A), and is significantly, though incompletely, predictive of ALDH18A1 mutations (Supplementary Fig. 3A). Dermal fibroblasts from Family FSP410 showed a decrease in proline biosynthesis by a stable isotope label substrate-loading test (Fig. 2B). The degree of observed residual flux as measured by the accumulation of labelled proline relative to the natural isotope is comparable to that measured in the first two patients reported to have biallelic ALDH18A1 mutations (40%; Baumgartner et al., 2005). However, the method we used differs from that report, and a direct comparison of the effect of different mutations needs to be performed for final conclusion. Regarding the pathogenic mechanisms in autosomal dominant patients, investigations are needed to directly compare the effects of homozygous and heterozygous mutations, especially to evaluate whether the latter leads to haploinsufficiency or a dominant negative gain-of-function. A previous report on a patient homozygous for a frameshift mutation resulting in no protein expression (Fischer et al., 2014) argues against haploinsufficiency as the pathogenic mechanism underlying dominant mutations.
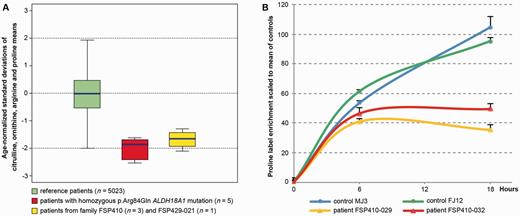
Plasma amino acid profiles and proline biosynthesis flux analysis. (A) Boxplots show the mean of four plasma amino acid levels (citrulline, ornithine, proline and arginine) expressed as their age-normalized standard deviations (y-axis; see ‘Materials and methods’ section) compared among three groups of patients: the full Necker hospital cohort of patients with no known metabolic disorders, aged >15 years (n = 5023; green); patients from the first family in which ALDH18A1 mutations were reported (n = 5; red); and the patients from Families FSP410 and FSP429 (this study) for which plasma amino acids were available (n = 4; yellow). The average of the four amino acid levels in ALDH18A1 mutated patients maps at approximately−2 SD. (B) Time course of stable isotope ratio (labelled versus natural) enrichment of proline reflecting the rate of proline biosynthesis from 13C5-labelled glutamine in fibroblasts. Two ALDH18A1-mutated patients (red and orange lines) are compared to two controls, i.e. members of the same family with no mutations (green and blue lines). Y-axis: label enrichment scaled to the mean of the controls at 18-h incubation. X-axis: hours of incubation in the presence of the tracer. Assuming a stationary phase at 18-h incubation time, proline biosynthesis in two patients harbouring ALDH18A1 p.Val120Ala mutation was estimated to be 42% of controls.
Furthermore, for autosomal recessive patients for which the amino acid profile was normal, another moonlighting, unknown, function of P5CS might be involved in the pathophysiology. Proline metabolism is a major actor in adaptation to osmotic stress and cellular redox stress (Hu et al., 2008a, b; Yasuda et al., 2013; Fichman et al., 2014). It is also involved in the apoptosis pathway; of note P5CS was shown to be upregulated by p53 (Hu et al., 2008b). Another field of interest is the mitochondria, as defects in mitochondrial structure have been described in PYCR1-linked neurocutaneous disease (Reversade et al., 2009). No such abnormalities had previously been identified in P5CS deficiency syndromes (Skidmore et al., 2011). Recently, Fischer et al. (2014) described swollen mitochondria in fibroblasts from their P5CS-deficient patient by electron microscopy on skin biopsies, but they could not observe them on cultured fibroblasts. In cultured fibroblasts from Patients FSP410‐29 and FSP410‐32, we found normal mitochondrial structure or membrane potential (data not shown). Renewed efforts are warranted to elucidate the cellular basis of ALDH18A1-related phenotypes.
Mitochondrial ornithine deficiency as a possible common scheme for multiple metabolic disorders
There are few metabolic causes of persistent hypocitrullinaemia after ruling out obvious and usually transient mechanisms of intestinal dysfunction: (i) urea cycle defects—notably, ornithine transcarbamylase (OTC), carbamoylphosphate synthase 1 (CPS1) and N-acetylglutamate synthetase (NAGS) deficiencies; (ii) ornithine translocase deficiency (or triple H syndrome); (iii) respiratory chain deficiencies (Rabier et al., 1998); and (iv) P5CS/ALDH18A1 deficiency (Baumgartner et al., 2000, 2005). Several biochemical hints allow for distinction between the enzymatic defects. Likewise, proline is decreased in P5CS deficiency only, orotic aciduria is present in OTC deficiency only and hyperammonemia is mainly observed in the fed state for urea cycle defects, whereas it manifests during fasting in P5CS deficiency. Decreased plasma citrulline levels could therefore be an excellent trait biomarker for ALDH18A1-linked autosomal dominant HSP, particularly as the clinical picture is not specific (relatively pure HSP form). Furthermore, the combination of low ornithine, citrulline, arginine and proline in plasma is significantly predictive of ALDH18A1-linked autosomal dominant HSP (Supplementary Fig. 3A).
The fact that the urea cycle defects and autosomal recessive HSP share common biomarkers as well as features of motor neuron degeneration is of interest and suggests that common metabolic mechanisms may be involved. Arginase deficiency (ARG1 mutations), an enzymatic step of the cytosolic part of the urea cycle (Fig. 3), is characterized by very high plasma levels of arginine and presumably decreased ornithine recycling. Triple H syndrome (SLC25A15 mutations), a defect of the translocase responsible for transporting ornithine from the cytosol into the mitochondria (Fig. 3), leads to high plasma levels of ornithine, thus reflecting low mitochondrial ornithine. Plasma citrulline is normal in arginase deficiency, but decreased in triple H syndrome. Both metabolic diseases are associated with HSP in adults (Sedel et al., 2007). Therefore, while plasma citrulline seems to be the most consistent biomarker of ALDH18A1-linked autosomal dominant HSP, a decrease in the mitochondrial pool of ornithine may be the common feature of arginase deficiency, triple H syndrome and P5CS deficiency (Fig. 3). We speculate that mitochondrial ornithine deficiency may be responsible for motor neuron degeneration in these diseases. This may have important therapeutic implications for patients with ALDH18A1-linked autosomal dominant HSP, because long-term supplementation in citrulline should be considered by analogy with its successful usage in patients with inborn errors of metabolism that directly or indirectly affect the urea cycle function—i.e. OTC deficiency or lysinuric protein intolerance (Haberle et al., 2012).
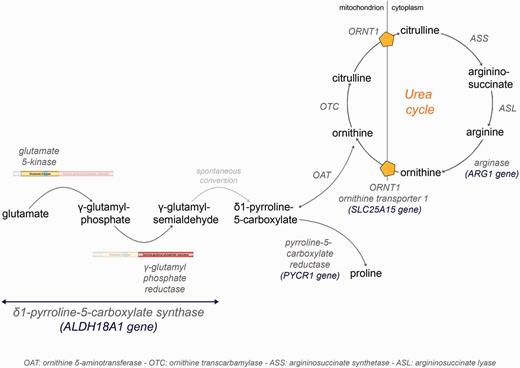
Delta-1-pyrroline-5-carboxylate synthase (P5CS) pathway. Schematic of the P5CS pathway. Enzyme-catalysed steps of proline and ornithine synthesis from glutamate, as well as urea cycle, are in dark grey. Mutations in ALDH18A1 and PYCR1 were previously reported in autosomal recessive neurocutaneous syndromes (Baumgartner et al., 2000; Reversade et al., 2009), with some pyramidal involvement. Deficiencies for arginase (ARG1 mutations) and ornithine transporter (SLC25A15 mutations, triple H syndrome) also share features of motor neuron degeneration (Sedel et al., 2007). We speculate that a decrease in the mitochondrial ornithine pool may be the common feature of the latter two and ALDH18A1-linked autosomal dominant HSP.
In conclusion, we describe seven novel mutations in ALDH18A1 in seven HSP pedigrees. Six patients from two families with complex autosomal recessive HSP broaden the phenotypic spectrum linked to this gene, with an emphasis on the pyramidal component and the absence of cutaneous signs. More importantly, we report ALDH18A1 mutations segregating in an autosomal dominant pattern in 17 patients from five pedigrees with pure or slightly complex HSP, thus adding a new gene involved in autosomal dominant HSP. We also describe the first potential plasma biomarker of autosomal dominant HSP that can be implemented in daily practice. Due to the great diversity of HSP, plasma biomarkers are invaluable diagnostic tools for the screening of certain genetic entities among all possible aetiologies of spastic paraplegia or to validate the causality of novel variants found in high throughput sequencing methods in genetic diagnosis. Likewise, plasma very long chain fatty acids and 25-/27-hydroxycholesterols are now established biomarkers of HSP, leading to more effective detection and dedicated care of adrenomyeloneuropathy (ABCD1 mutations) and SPG5 (CYP7B1 mutations) (Schule et al., 2010; Kemp et al., 2012). Provided that our biochemical findings are validated in a larger population of ALDH18A1-linked autosomal dominant HSP patients, we recommend the use of plasma amino acid chromatography as part of diagnostic work-up in HSP. These results are also illustrative of the notion of ‘genetics remodelling’ brought by Next Generation Sequencing, as has been demonstrated for other genes (Esteves et al., 2014; Synofzik et al., 2014; Tesson et al., 2015). The questioning of assumed transmission modes and new delineations in phenotype-genotype correlations are hallmarks of that field. Finally, we confirm that the metabolism of amino acids is an important cellular pathway in HSPs, which may help in the understanding of their physiopathology.
Abbreviations
Acknowledgements
The authors are grateful to the patients, to Prs G. Ponsot, A. David, J-Ph. Neau, V. Paquis and Drs M. Clanet, P. Heine, A. Jacquette, L. Freemann, R. De Paz, M.L Monin and M. Aupy, for referring patients, to Justine Guégan, Dr C. Nava and Dr I. Moszer for Bioinformatic analysis, to Wassila Carpentier for SNP genotyping, to Sarah Boster for manuscript proofreading, to the Genotyping and Sequencing facility and the DNA and cell bank of the ICM for sample preparation and processing. We thank the Centre de Référence des Maladies Métaboliques (Necker Hospital, Paris) for sharing biochemical data of their cohort of patients.
Funding
The authors’ work was partially funded by the Agence Nationale de la Recherche (SPATAX-QUEST, to G.S.), the Fundação para a Ciência e a Tecnologia (FCT-ANR/BEX-GMG/0008/2013, to I.A.), the European Community’s Seventh Framework Programme (FP7/2007‐2013) under grant agreement n° 2012‐305121 ‘Integrated European –omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS, to A.B., G.S. and A.D.)’, the European Union ERA-Net for Research Programmes on Rare Diseases (NEUROLIPID, to G.S.), the VERUM Foundation (to A.B., A.D. and G.S.), the NIH (R01NS075764, 5R01NS072248 and U54NS065712 to S.Z.), the Fondation Lejeune (to C.O.) and benefited from the program ‘Investissements d’Avenir’ ANR-10-IAIHU-06 (to the ICM). M.C. was the recipient of a fellowship from the Fonds de la Recherche Scientifique (aspirant FNRS). S.M. was the recipient of a fellowship (SFRH/BD/87189/2012) from the FCT (Fundação para a Ciência e Tecnologia) supported by POPH/FSE funding.
Supplementary material
Supplementary material is available at Brain online.
References
Author notes
#Present address: Department of Neurology, Oslo University Hospital; and Faculty of Medicine, Oslo University, Oslo, Norway


{kind=link}
{kind=link}
{kind=link}
{kind=link}