




Abstract
Additional neurological features have recently been described in seven families transmitting pathogenic mutations in OPA1, the most common cause of autosomal dominant optic atrophy. However, the frequency of these syndromal ‘dominant optic atrophy plus’ variants and the extent of neurological involvement have not been established. In this large multi-centre study of 104 patients from 45 independent families, including 60 new cases, we show that extra-ocular neurological complications are common in OPA1 disease, and affect up to 20% of all mutational carriers. Bilateral sensorineural deafness beginning in late childhood and early adulthood was a prominent manifestation, followed by a combination of ataxia, myopathy, peripheral neuropathy and progressive external ophthalmoplegia from the third decade of life onwards. We also identified novel clinical presentations with spastic paraparesis mimicking hereditary spastic paraplegia, and a multiple sclerosis-like illness. In contrast to initial reports, multi-system neurological disease was associated with all mutational subtypes, although there was an increased risk with missense mutations [odds ratio = 3.06, 95% confidence interval = 1.44–6.49; P = 0.0027], and mutations located within the guanosine triphosphate-ase region (odds ratio = 2.29, 95% confidence interval = 1.08–4.82; P = 0.0271). Histochemical and molecular characterization of skeletal muscle biopsies revealed the presence of cytochrome c oxidase-deficient fibres and multiple mitochondrial DNA deletions in the majority of patients harbouring OPA1 mutations, even in those with isolated optic nerve involvement. However, the cytochrome c oxidase-deficient load was over four times higher in the dominant optic atrophy + group compared to the pure optic neuropathy group, implicating a causal role for these secondary mitochondrial DNA defects in disease pathophysiology. Individuals with dominant optic atrophy plus phenotypes also had significantly worse visual outcomes, and careful surveillance is therefore mandatory to optimize the detection and management of neurological disability in a group of patients who already have significant visual impairment.
Introduction
Mitochondria supply most of the cellular adenosine triphosphate (ATP) requirements through oxidative phosphorylation, a process that involves five protein complexes located along the inner mitochondrial membrane. Mitochondrial DNA (mtDNA) codes for 13 of the key structural polypeptides that constitute these multimeric complexes, in addition to two ribosomal RNAs and 22 transfer RNAs which are required for initiating translation and protein synthesis (DiMauro and Schon, 2003; Schapira, 2006). Over the past decade, a growing number of nuclear genetic defects have been identified that disrupt mitochondrial function either by a reduction in mtDNA copy number, and/or the accumulation of secondary mtDNA deletions: POLG1, POLG2, PEO1, SLC25A4, TYMP, DGUOK, TK2, SUCLA2, SUCLG1, MPV17 and RRM2B (Hudson and Chinnery, 2006; Chinnery and Zeviani, 2008; Copeland, 2008). These disorders of mtDNA maintenance constitute an important group of human genetic diseases and they manifest as an array of clinical syndromes ranging from severe infantile encephalomyopathy, hepatic failure and epilepsy, to late-onset ataxia, peripheral neuropathy and myopathy, or isolated progressive external ophthalmoplegia. However, visual failure is rare in this group of patients.
A recent unexpected addition to this list of genes linked to nuclear mitochondrial disorders was OPA1, the major causative gene in ∼60% of families with autosomal dominant optic atrophy (DOA, OMIM 605290) (Alexander et al., 2000; Delettre et al., 2000; Yu-Wai-Man et al., 2009a). OPA1 consists of 30 exons spanning 100 kb of genomic DNA on the long arm of chromosome 3 (3q28–q29), and the protein product is a 960 amino acid polypeptide which co-localizes to the inner mitochondrial membrane (Davies and Votruba, 2006). OPA1 contains a highly conserved functional GTPase domain shared by members of the dynamin superfamily of mechanoenzymes, and it regulates several important cellular processes including the stability of the mitochondrial network via its pro-fusion properties (Chan, 2007), mitochondrial bioenergetic output through a postulated effect on the assembly and stability of complex I and IV subunits (Chevrollier et al., 2008; Zanna et al., 2008), and the sequestration of pro-apoptotic cytochrome c molecules within the mitochondrial cristae spaces (Cipolat et al., 2006; Frezza et al., 2006).
DOA is one of the most common inherited optic neuropathies and it classically presents in early childhood with progressive visual failure, with selective retinal ganglion cell loss as its defining pathological characteristic (Newman and Biousse, 2004; Yu-Wai-Man et al., 2009a). However, even in the pre-molecular era, patients with DOA and other clinical manifestations, most commonly sensorineural deafness, ataxia, myopathy and progressive external ophthalmoplegia, were recognized (Hoyt, 1980; Meire et al., 1985). The significance of these syndromal DOA variants was recently highlighted by the identification of cytochrome c oxidase (COX)-deficient muscle fibres and multiple mtDNA deletions in skeletal muscle biopsies from these patients, implicating a previously unsuspected role of OPA1 in mtDNA maintenance (Amati-Bonneau et al., 2008; Hudson et al., 2008; Zeviani, 2008). Although intriguing from a mechanistic perspective, the broader clinical significance of our observations was unclear, given that only seven families had been identified. We therefore conducted a systematic clinical and molecular study of OPA1 positive families to establish the frequency of DOA plus syndrome (DOA+) in OPA1 disease, describe the phenotypic spectrum and the natural history of these additional neuromuscular features, and define any genotype–phenotype correlations.
Materials and methods
Patient cohorts
OPA1 positive families included in this study were identified from the following groups. In addition, we conducted a systematic review of the literature to identify other DOA+ families with confirmed OPA1 mutations. These published data were included in our final analysis to provide a definitive description of the visual prognosis and genotype–phenotype associations seen in DOA+.
Our initial cohort was assembled as part of an ongoing epidemiological study of inherited optic neuropathies in the North of England. OPA1 screening was performed on probands with a clinical diagnosis of DOA where previous investigations had excluded other compressive, inflammatory and infiltrative causes for their bilateral optic atrophy. Additional family members were examined both during the initial diagnostic work up of the proband, and at the time of active contact tracing following the identification of a pathogenic OPA1 mutation.
The relatively high prevalence of DOA+ phenotypes among patients with OPA1 mutations in the North of England prompted us to extend our original study to other diagnostic and research centres with an interest in neuro-ophthalmological disorders. DNA samples were analysed for patients with a high suspicion of an underlying mitochondrial genetic disease due to the multi-system pattern of their clinical presentations, evidence of a mitochondrial biochemical defect, and/or the presence of multiple mtDNA deletions. Visual failure was a prominent feature of their phenotypes and when initial screens for POLG1, POLG2, PEO1 and SLC25A4 were found to be negative, OPA1 sequencing was performed.
Phenotypic evaluation
All patients harbouring pathogenic OPA1 mutations were comprehensively assessed by a team of experienced ophthalmologists and neurologists to define both the extent and the severity of their ocular and neuromuscular deficits. A complete orthoptic assessment was performed to document any abnormalities of ocular motility, supplemented by formal eye-movement recordings where appropriate. Ancillary investigations including visual electrophysiology, peripheral nerve conduction studies, electromyography, electroencephalography, audiometry and neuroimaging were also performed as dictated by the patient’s symptoms and the familial phenotype to confirm possible cases of non-penetrance.
Best corrected visual acuity was measured using the Snellen chart and for the purpose of statistical analysis, Snellen ratios were converted to LogMAR (logarithm of the minimum angle of resolution) decimal values. A LogMAR value of 0 is equivalent to 6/6 Snellen vision and a value of 1.0 is equivalent to 6/60 Snellen vision, the largest optotype on standard Snellen charts. Patients with visual acuities reduced to counting fingers were assigned a LogMAR value of 2.0, whilst those with only hand movement perception were given a LogMAR value of 2.3 (Schulze-Bonsel et al., 2006; Lange et al., 2009).
Identification of OPA1 mutations
The entire OPA1-coding region and flanking exon–intron boundaries were amplified by the polymerase chain reaction (PCR) and sequenced with BigDyeTM terminator cycle chemistries on an appropriate genetic analyser (Primers and cycling conditions are available on request). Sequence chromatograms were compared with the Genbank OPA1 reference sequence (Accession number AB011139) and all genetic variants were confirmed by reverse sequencing. Novel OPA1 mutations not reported on the eOPA1 database (http://lbbma.univ-angers.fr/eOPA1/) were screened in a panel of 150–300 age-matched controls using (i) a multiplex primer extension assay and product analysis with matrix-associated laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) (Sequenom, San Diego, CA); or (ii) BigDyeTM sequencing of the relevant PCR-amplified OPA1 fragments (Stewart et al., 2008).
Reverse-transcriptase PCR
Total RNA was extracted from blood samples with TrizolTM reagent (Invitrogen, Groningen, The Netherlands), and complementary DNA was obtained using SuperScriptTM RNA polymerase and poly-T as the primer (Invitrogen, Cergy Pontoise, France). PCR was performed with the following primer pair: (i) forward 5′-ACG CAA GAT CAT CTG CCA CGG-3′; and (ii) reverse 5′-CCT GCT TGG ACT GGC TAC A-3′, to amplify a 597 bp fragment overlapping OPA1 exons 9–14 (Amati-Bonneau et al., 2004). The generated products were analysed on a 1% agarose gel and sequenced with BigDyeTM terminator cycle chemistries.
Mitochondrial histochemistry
Skeletal muscle biopsies collected from 34 OPA1 mutation carriers were snap frozen in melting liquid isopentane (–150°C) and then stored at –80°C. The blocks were serially sectioned on a cryostat before being stained for COX, succinate dehydrogenase (SDH) and sequential COX–SDH activities, using standard histochemical protocols (Johnson and Barron, 1996). The proportion of muscle fibres exhibiting a mitochondrial oxidative phosphorylation defect can be accurately quantified, as normal COX-positive fibres stain brown; whereas COX-negative fibres are highlighted blue as a result of impaired complex IV activity. The histochemical findings in our DOA+ cohort were compared with an age-matched control group of 20 skeletal muscle biopsies from healthy subjects (mean age = 51.1 years, SD = 10.7 years, P = 0.4334) (Brierley et al., 1996).
Mitochondrial DNA deletion and depletion assays
Multiple mtDNA deletions in homogenate skeletal muscle DNA were identified by Southern blot and/or long-range PCR (Amati-Bonneau et al., 2008; Hudson et al., 2008). mtDNA copy number was determined using a well-established iQ SYBR Green protocol on the MyiQTM real-time PCR detection system (Biorad, USA), with MTND1 as the mtDNA reference gene and GAPDH as the nuclear DNA reference gene (Pyle et al., 2007; Cree et al., 2008). Both assays were optimized and confirmed to be linear over an appropriate concentration range by the standard curve method, and all measurements were done in triplicate. The relative mtDNA copy number was derived using the 2(2–ΔCt) equation, where ΔCt was the difference in threshold cycle value (Ct) between MTND1 and GAPDH, and accounting for two copies of GAPDH per cell nucleus. mtDNA copy number in skeletal muscle homogenate samples was also determined for a group of age-matched healthy controls (mean age = 43.1 years, SD = 20.1 years, n = 20, P = 0.1300), and paediatric patients with known mtDNA depletion syndromes (mean age = 3.3 years, SD = 2.7 years, n = 6, P < 0.0001).
Statistical analysis
Statistical analysis was performed using GraphPadTM v.4 statistical software (San Diego, CA). Fisher’s exact test and an independent sample t-test were used for group comparisons, as required. Prevalence figures were derived from the mid-2008 census data for the North of England, and 95% CIs were calculated using Poisson distribution variables for small numbers (Bland, 2000).
Results
OPA1 cohorts
The epidemiological study identified OPA1 mutations in 14 genetically distinct OPA1 families, and a total of 64 affected family members were living in the North of England. Their mean age was 39.5 years (SD = 17.8 years, range 6.0–78.0 years), with a male to female ratio of 1.06:1, and DOA+ clinical features were present in 11 out of 64 patients (17.2%) from 6 out of 14 (42.9%) families. In collaboration with other clinical centres, 49 additional DOA+ patients from 21 families harbouring OPA1 mutations were also found: British, n = 8; French, n = 5; Italian, n = 3; German, n = 2; Norwegian, n = 1; Austrian, n = 1; and Brazilian, n = 1 (mean age = 44.9 years, SD = 18.4 years). A systematic review of the literature revealed 18 published OPA1 positive families segregating a DOA+ phenotype and clinical information was available for 44 affected individuals (mean age = 43.5 years, SD = 17.0 years, Table 1).
Clinical and molecular characteristics of patients with DOA+ phenotypes
| Pedigree | Age (years) | Sex | FHx | Clinical features | OPA1 mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Optic atrophy | Deafness | Ataxia | Myopathy | Neuropathy | PEO | Others | Complementary DNA | Amino acid change | Domain | ||||
| UK-1 | 54 | M | + | + | + | c.1212 + 3a>t | – | – | |||||
| UK-2 | 36 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-2 | 55 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-3 | 59 | M | + | + | + | HSP Migraine | c.876–878del(TGT) | p.V294fsX667 | – | ||||
| UK-3 | 75a | M | + | + | HSP | c.876–878del(TGT) | p.V294fsX667 | – | |||||
| UK-4 | 44 | M | + | + | + | c.1198C>T | p.P400S | GTPase | |||||
| UK-5 | 48 | F | + | + | + | HSP | c.889C>T | p.Q297X | – | ||||
| UK-5 | 21 | M | + | + | HSP | c.889C>T | p.Q297X | – | |||||
| UK-6 | 58 | F | + | + | + | MS | c.2613 + 1g>a | – | – | ||||
| UK-7 | 46 | F | + | + | c.2708_2711del(TTAG) | p.V903fsX3 | – | ||||||
| UK-8 | 44 | F | + | + | + | + | Diabetes | c.32 + 1g>a | – | – | |||
| UK-8 | 53 | M | + | + | + | Diabetes | c.32 + 1g>a | – | – | ||||
| UK-9 | 15 | M | + | + | + | + | c.1212 + 1G>A | – | – | ||||
| UK-10 | 42 | F | + | + | c.1202G>A | p.G401D | GTPase | ||||||
| UK-11 | 70a | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-11 | 64 | F | + | + | + | + | + | + | + | Diabetes | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 42 | M | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| UK-11 | 38 | F | + | + | + | + | + | + | + | Migraine | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-12 | 43 | M | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 44 | F | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 69 | F | + | + | c.1294A>G | p.I432V | GTPase | ||||||
| UK-13 | 43 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| UK-14 | 44 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 42 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 73 | M | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| AU-1 | 35 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 34 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 61 | F | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| BE-1 | 72 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 59 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 29 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 25 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BR-1 | 9 | M | + | + | + | CAPOS | c.1327G>A | p.A443T | GTPase | ||||
| CN-1 | 46 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-1 | 22 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-2 | 34 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 36 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 33 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 29 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-3 | 61 | F | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 55 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 37 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 24 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| DE-1 | 14 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-1 | 13 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-2 | 11 | M | + | + | + | c.1313A>T | p.D438V | GTPase | |||||
| ES-1 | 57 | F | + | + | + | + | + | + | Dementia | c.1334G>A | p.R445H | GTPase | |
| ES-1 | 30 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FI-1 | F | + | + | + | c.970_978del(9) | p.R324_P326del | – | ||||||
| FR-1 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| FR-2 | 35 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| FR-3 | 14 | M | b | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-4 | 39 | M | + | + | + | + | + | c.728T>A | p.L243X | – | |||
| FR-5 | 53 | M | + | Epilepsy | c.1071A>G | p.A358Ac | – | ||||||
| FR-6 | 12 | F | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-7 | 20 | M | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-8 | 38 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-9 | 43 | M | + | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | |
| FR-10 | 67 | M | + | + | + | + | + | + | c.1069G>A | p.A357T | GTPase | ||
| FR-11 | 44 | M | + | MS | c.1937C>T | p.S646L | Dynamin | ||||||
| IT-1 | 64 | F | + | + | + | + | + | c.1462G>A | p.G488R | GTPase | |||
| IT-1 | 64a | M | + | + | + | + | + | + | Dementia | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 36a | M | + | + | + | Migraine, epilepsy | c.1462 A>G | p.G488R | GTPase | ||||
| IT-1 | 72 | F | + | + | + | + | + | + | c.1462 A>G | p.G488R | GTPase | ||
| IT-1 | 69 | M | + | + | + | + | + | + | Migraine | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 46 | F | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 44 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 53 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-2 | 12 | F | + | + | + | c.344C>T | p.A115V | Basic | |||||
| IT-3 | 69 | M | + | + | + | + | + | c.1484C>T | p.A495V | GTPase | |||
| IT-3 | 60 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 62 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 39 | F | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 68 | M | + | + | + | + | c.1484C>T | p.A495V | GTPase | ||||
| IT-4 | 38 | M | + | + | + | + | + | + | + | c.1316G>T | p.G439V | GTPase | |
| IT-4 | 6 | F | + | + | + | c.1316G>T | p.G439V | GTPase | |||||
| IT-5 | 59 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-5 | 53 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-6 | 47 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-6 | 46 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-7 | 42 | M | + | + | + | + | c.1745A>G | p.Y582C | Dynamin | ||||
| NO-1 | 60 | M | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 64 | F | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 43 | F | + | c.768C>G | p.S256R | CC | |||||||
| NO-1 | 35 | F | + | c.768C>G | p.S256R | CC | |||||||
| US-1 | 71 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 68 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 56 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 77 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 49 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 45 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 38 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 42 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 43 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 51 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 26 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 19 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-2 | 48 | F | + | + | + | c113_130del(18) | p.R38_S43del | – | |||||
| Pedigree | Age (years) | Sex | FHx | Clinical features | OPA1 mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Optic atrophy | Deafness | Ataxia | Myopathy | Neuropathy | PEO | Others | Complementary DNA | Amino acid change | Domain | ||||
| UK-1 | 54 | M | + | + | + | c.1212 + 3a>t | – | – | |||||
| UK-2 | 36 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-2 | 55 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-3 | 59 | M | + | + | + | HSP Migraine | c.876–878del(TGT) | p.V294fsX667 | – | ||||
| UK-3 | 75a | M | + | + | HSP | c.876–878del(TGT) | p.V294fsX667 | – | |||||
| UK-4 | 44 | M | + | + | + | c.1198C>T | p.P400S | GTPase | |||||
| UK-5 | 48 | F | + | + | + | HSP | c.889C>T | p.Q297X | – | ||||
| UK-5 | 21 | M | + | + | HSP | c.889C>T | p.Q297X | – | |||||
| UK-6 | 58 | F | + | + | + | MS | c.2613 + 1g>a | – | – | ||||
| UK-7 | 46 | F | + | + | c.2708_2711del(TTAG) | p.V903fsX3 | – | ||||||
| UK-8 | 44 | F | + | + | + | + | Diabetes | c.32 + 1g>a | – | – | |||
| UK-8 | 53 | M | + | + | + | Diabetes | c.32 + 1g>a | – | – | ||||
| UK-9 | 15 | M | + | + | + | + | c.1212 + 1G>A | – | – | ||||
| UK-10 | 42 | F | + | + | c.1202G>A | p.G401D | GTPase | ||||||
| UK-11 | 70a | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-11 | 64 | F | + | + | + | + | + | + | + | Diabetes | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 42 | M | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| UK-11 | 38 | F | + | + | + | + | + | + | + | Migraine | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-12 | 43 | M | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 44 | F | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 69 | F | + | + | c.1294A>G | p.I432V | GTPase | ||||||
| UK-13 | 43 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| UK-14 | 44 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 42 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 73 | M | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| AU-1 | 35 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 34 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 61 | F | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| BE-1 | 72 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 59 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 29 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 25 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BR-1 | 9 | M | + | + | + | CAPOS | c.1327G>A | p.A443T | GTPase | ||||
| CN-1 | 46 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-1 | 22 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-2 | 34 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 36 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 33 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 29 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-3 | 61 | F | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 55 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 37 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 24 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| DE-1 | 14 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-1 | 13 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-2 | 11 | M | + | + | + | c.1313A>T | p.D438V | GTPase | |||||
| ES-1 | 57 | F | + | + | + | + | + | + | Dementia | c.1334G>A | p.R445H | GTPase | |
| ES-1 | 30 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FI-1 | F | + | + | + | c.970_978del(9) | p.R324_P326del | – | ||||||
| FR-1 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| FR-2 | 35 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| FR-3 | 14 | M | b | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-4 | 39 | M | + | + | + | + | + | c.728T>A | p.L243X | – | |||
| FR-5 | 53 | M | + | Epilepsy | c.1071A>G | p.A358Ac | – | ||||||
| FR-6 | 12 | F | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-7 | 20 | M | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-8 | 38 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-9 | 43 | M | + | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | |
| FR-10 | 67 | M | + | + | + | + | + | + | c.1069G>A | p.A357T | GTPase | ||
| FR-11 | 44 | M | + | MS | c.1937C>T | p.S646L | Dynamin | ||||||
| IT-1 | 64 | F | + | + | + | + | + | c.1462G>A | p.G488R | GTPase | |||
| IT-1 | 64a | M | + | + | + | + | + | + | Dementia | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 36a | M | + | + | + | Migraine, epilepsy | c.1462 A>G | p.G488R | GTPase | ||||
| IT-1 | 72 | F | + | + | + | + | + | + | c.1462 A>G | p.G488R | GTPase | ||
| IT-1 | 69 | M | + | + | + | + | + | + | Migraine | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 46 | F | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 44 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 53 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-2 | 12 | F | + | + | + | c.344C>T | p.A115V | Basic | |||||
| IT-3 | 69 | M | + | + | + | + | + | c.1484C>T | p.A495V | GTPase | |||
| IT-3 | 60 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 62 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 39 | F | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 68 | M | + | + | + | + | c.1484C>T | p.A495V | GTPase | ||||
| IT-4 | 38 | M | + | + | + | + | + | + | + | c.1316G>T | p.G439V | GTPase | |
| IT-4 | 6 | F | + | + | + | c.1316G>T | p.G439V | GTPase | |||||
| IT-5 | 59 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-5 | 53 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-6 | 47 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-6 | 46 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-7 | 42 | M | + | + | + | + | c.1745A>G | p.Y582C | Dynamin | ||||
| NO-1 | 60 | M | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 64 | F | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 43 | F | + | c.768C>G | p.S256R | CC | |||||||
| NO-1 | 35 | F | + | c.768C>G | p.S256R | CC | |||||||
| US-1 | 71 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 68 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 56 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 77 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 49 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 45 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 38 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 42 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 43 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 51 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 26 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 19 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-2 | 48 | F | + | + | + | c113_130del(18) | p.R38_S43del | – | |||||
AU = Austria; BE = Belgium; BR = Brazil; CN = China; DE = Germany; ES = Spain; FI = Finland; FR = France; IT = Italy; NO = Norway; UK = United Kingdom; US = USA; AA = amino acid; FHx = family history; HSP = hereditary spastic paraplegia; MS = multiple sclerosis-like illness; CAPOS = cerebellar ataxia, areflexia, pes cavus, optic atrophy and sensorineural hearing loss; CC = coiled-coil domain; GE = GTPase effector domain; PEO = progressive external ophthalmoplegia.
aAge of death.
bDe novo OPA1 mutation.
cIn-frame skipping of exon 12 (p.T381-N404del).
BE-1: (Meire et al., 1985; Payne et al., 2004); CN-1: (Li et al., 2005); CN-2: (Ke et al., 2006); CN-3: (Chen et al., 2007); ES-1: (Amati-Bonneau et al., 2005); FI-1: (Puomila et al., 2005); FR-6, FR-7, FR-8:(Amati-Bonneau et al., 2005); FR-9, FR-10: (Amati-Bonneau et al., 2008); IT-4: (Amati-Bonneau et al., 2008; Liguori et al., 2008); IT-5: (Amati-Bonneau et al., 2008); IT-6: (Spinazzi et al., 2008); IT-7: (Ferraris et al., 2008); UK-11: (Hudson et al., 2008); UK-12, UK-13, UK-14: (Stewart et al., 2008); US-1: (Payne et al., 2004); US-2: (Milone et al., 2009).
Clinical and molecular characteristics of patients with DOA+ phenotypes
| Pedigree | Age (years) | Sex | FHx | Clinical features | OPA1 mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Optic atrophy | Deafness | Ataxia | Myopathy | Neuropathy | PEO | Others | Complementary DNA | Amino acid change | Domain | ||||
| UK-1 | 54 | M | + | + | + | c.1212 + 3a>t | – | – | |||||
| UK-2 | 36 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-2 | 55 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-3 | 59 | M | + | + | + | HSP Migraine | c.876–878del(TGT) | p.V294fsX667 | – | ||||
| UK-3 | 75a | M | + | + | HSP | c.876–878del(TGT) | p.V294fsX667 | – | |||||
| UK-4 | 44 | M | + | + | + | c.1198C>T | p.P400S | GTPase | |||||
| UK-5 | 48 | F | + | + | + | HSP | c.889C>T | p.Q297X | – | ||||
| UK-5 | 21 | M | + | + | HSP | c.889C>T | p.Q297X | – | |||||
| UK-6 | 58 | F | + | + | + | MS | c.2613 + 1g>a | – | – | ||||
| UK-7 | 46 | F | + | + | c.2708_2711del(TTAG) | p.V903fsX3 | – | ||||||
| UK-8 | 44 | F | + | + | + | + | Diabetes | c.32 + 1g>a | – | – | |||
| UK-8 | 53 | M | + | + | + | Diabetes | c.32 + 1g>a | – | – | ||||
| UK-9 | 15 | M | + | + | + | + | c.1212 + 1G>A | – | – | ||||
| UK-10 | 42 | F | + | + | c.1202G>A | p.G401D | GTPase | ||||||
| UK-11 | 70a | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-11 | 64 | F | + | + | + | + | + | + | + | Diabetes | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 42 | M | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| UK-11 | 38 | F | + | + | + | + | + | + | + | Migraine | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-12 | 43 | M | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 44 | F | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 69 | F | + | + | c.1294A>G | p.I432V | GTPase | ||||||
| UK-13 | 43 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| UK-14 | 44 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 42 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 73 | M | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| AU-1 | 35 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 34 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 61 | F | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| BE-1 | 72 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 59 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 29 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 25 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BR-1 | 9 | M | + | + | + | CAPOS | c.1327G>A | p.A443T | GTPase | ||||
| CN-1 | 46 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-1 | 22 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-2 | 34 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 36 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 33 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 29 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-3 | 61 | F | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 55 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 37 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 24 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| DE-1 | 14 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-1 | 13 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-2 | 11 | M | + | + | + | c.1313A>T | p.D438V | GTPase | |||||
| ES-1 | 57 | F | + | + | + | + | + | + | Dementia | c.1334G>A | p.R445H | GTPase | |
| ES-1 | 30 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FI-1 | F | + | + | + | c.970_978del(9) | p.R324_P326del | – | ||||||
| FR-1 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| FR-2 | 35 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| FR-3 | 14 | M | b | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-4 | 39 | M | + | + | + | + | + | c.728T>A | p.L243X | – | |||
| FR-5 | 53 | M | + | Epilepsy | c.1071A>G | p.A358Ac | – | ||||||
| FR-6 | 12 | F | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-7 | 20 | M | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-8 | 38 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-9 | 43 | M | + | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | |
| FR-10 | 67 | M | + | + | + | + | + | + | c.1069G>A | p.A357T | GTPase | ||
| FR-11 | 44 | M | + | MS | c.1937C>T | p.S646L | Dynamin | ||||||
| IT-1 | 64 | F | + | + | + | + | + | c.1462G>A | p.G488R | GTPase | |||
| IT-1 | 64a | M | + | + | + | + | + | + | Dementia | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 36a | M | + | + | + | Migraine, epilepsy | c.1462 A>G | p.G488R | GTPase | ||||
| IT-1 | 72 | F | + | + | + | + | + | + | c.1462 A>G | p.G488R | GTPase | ||
| IT-1 | 69 | M | + | + | + | + | + | + | Migraine | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 46 | F | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 44 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 53 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-2 | 12 | F | + | + | + | c.344C>T | p.A115V | Basic | |||||
| IT-3 | 69 | M | + | + | + | + | + | c.1484C>T | p.A495V | GTPase | |||
| IT-3 | 60 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 62 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 39 | F | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 68 | M | + | + | + | + | c.1484C>T | p.A495V | GTPase | ||||
| IT-4 | 38 | M | + | + | + | + | + | + | + | c.1316G>T | p.G439V | GTPase | |
| IT-4 | 6 | F | + | + | + | c.1316G>T | p.G439V | GTPase | |||||
| IT-5 | 59 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-5 | 53 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-6 | 47 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-6 | 46 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-7 | 42 | M | + | + | + | + | c.1745A>G | p.Y582C | Dynamin | ||||
| NO-1 | 60 | M | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 64 | F | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 43 | F | + | c.768C>G | p.S256R | CC | |||||||
| NO-1 | 35 | F | + | c.768C>G | p.S256R | CC | |||||||
| US-1 | 71 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 68 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 56 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 77 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 49 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 45 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 38 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 42 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 43 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 51 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 26 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 19 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-2 | 48 | F | + | + | + | c113_130del(18) | p.R38_S43del | – | |||||
| Pedigree | Age (years) | Sex | FHx | Clinical features | OPA1 mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Optic atrophy | Deafness | Ataxia | Myopathy | Neuropathy | PEO | Others | Complementary DNA | Amino acid change | Domain | ||||
| UK-1 | 54 | M | + | + | + | c.1212 + 3a>t | – | – | |||||
| UK-2 | 36 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-2 | 55 | F | + | + | + | + | Migraine | c.870 + 5g>a | – | – | |||
| UK-3 | 59 | M | + | + | + | HSP Migraine | c.876–878del(TGT) | p.V294fsX667 | – | ||||
| UK-3 | 75a | M | + | + | HSP | c.876–878del(TGT) | p.V294fsX667 | – | |||||
| UK-4 | 44 | M | + | + | + | c.1198C>T | p.P400S | GTPase | |||||
| UK-5 | 48 | F | + | + | + | HSP | c.889C>T | p.Q297X | – | ||||
| UK-5 | 21 | M | + | + | HSP | c.889C>T | p.Q297X | – | |||||
| UK-6 | 58 | F | + | + | + | MS | c.2613 + 1g>a | – | – | ||||
| UK-7 | 46 | F | + | + | c.2708_2711del(TTAG) | p.V903fsX3 | – | ||||||
| UK-8 | 44 | F | + | + | + | + | Diabetes | c.32 + 1g>a | – | – | |||
| UK-8 | 53 | M | + | + | + | Diabetes | c.32 + 1g>a | – | – | ||||
| UK-9 | 15 | M | + | + | + | + | c.1212 + 1G>A | – | – | ||||
| UK-10 | 42 | F | + | + | c.1202G>A | p.G401D | GTPase | ||||||
| UK-11 | 70a | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-11 | 64 | F | + | + | + | + | + | + | + | Diabetes | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 42 | M | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| UK-11 | 38 | F | + | + | + | + | + | + | + | Migraine | c.1635C>G | p.S545R | Dynamin |
| UK-11 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| UK-12 | 43 | M | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 44 | F | + | + | + | + | + | + | c.1294A>G | p.I432V | GTPase | ||
| UK-12 | 69 | F | + | + | c.1294A>G | p.I432V | GTPase | ||||||
| UK-13 | 43 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| UK-14 | 44 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 42 | F | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| UK-14 | 73 | M | + | + | + | + | + | c.1334G>A | p.R445H | GTPase | |||
| AU-1 | 35 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 34 | F | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| AU-1 | 61 | F | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||||
| BE-1 | 72 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 59 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 29 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BE-1 | 25 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| BR-1 | 9 | M | + | + | + | CAPOS | c.1327G>A | p.A443T | GTPase | ||||
| CN-1 | 46 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-1 | 22 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| CN-2 | 34 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 36 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 33 | M | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-2 | 29 | F | + | + | + | c.1202G>A | p.G401D | GTPase | |||||
| CN-3 | 61 | F | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 55 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 37 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| CN-3 | 24 | M | + | + | + | c.2848_2849del(GA) | p.D950fsX4 | – | |||||
| DE-1 | 14 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-1 | 13 | F | + | + | + | c.635_636del(AA) | p.K212fsX4 | – | |||||
| c.1642G>A | p.V548I | Dynamin | |||||||||||
| DE-2 | 11 | M | + | + | + | c.1313A>T | p.D438V | GTPase | |||||
| ES-1 | 57 | F | + | + | + | + | + | + | Dementia | c.1334G>A | p.R445H | GTPase | |
| ES-1 | 30 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FI-1 | F | + | + | + | c.970_978del(9) | p.R324_P326del | – | ||||||
| FR-1 | 30 | M | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | ||
| FR-2 | 35 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| FR-3 | 14 | M | b | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-4 | 39 | M | + | + | + | + | + | c.728T>A | p.L243X | – | |||
| FR-5 | 53 | M | + | Epilepsy | c.1071A>G | p.A358Ac | – | ||||||
| FR-6 | 12 | F | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-7 | 20 | M | + | + | c.1334G>A | p.R445H | GTPase | ||||||
| FR-8 | 38 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| FR-9 | 43 | M | + | + | + | + | + | + | + | c.1635C>G | p.S545R | Dynamin | |
| FR-10 | 67 | M | + | + | + | + | + | + | c.1069G>A | p.A357T | GTPase | ||
| FR-11 | 44 | M | + | MS | c.1937C>T | p.S646L | Dynamin | ||||||
| IT-1 | 64 | F | + | + | + | + | + | c.1462G>A | p.G488R | GTPase | |||
| IT-1 | 64a | M | + | + | + | + | + | + | Dementia | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 36a | M | + | + | + | Migraine, epilepsy | c.1462 A>G | p.G488R | GTPase | ||||
| IT-1 | 72 | F | + | + | + | + | + | + | c.1462 A>G | p.G488R | GTPase | ||
| IT-1 | 69 | M | + | + | + | + | + | + | Migraine | c.1462 A>G | p.G488R | GTPase | |
| IT-1 | 46 | F | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 44 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-1 | 53 | M | + | + | + | c.1462 A>G | p.G488R | GTPase | |||||
| IT-2 | 12 | F | + | + | + | c.344C>T | p.A115V | Basic | |||||
| IT-3 | 69 | M | + | + | + | + | + | c.1484C>T | p.A495V | GTPase | |||
| IT-3 | 60 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 62 | M | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 39 | F | + | + | + | c.1484C>T | p.A495V | GTPase | |||||
| IT-3 | 68 | M | + | + | + | + | c.1484C>T | p.A495V | GTPase | ||||
| IT-4 | 38 | M | + | + | + | + | + | + | + | c.1316G>T | p.G439V | GTPase | |
| IT-4 | 6 | F | + | + | + | c.1316G>T | p.G439V | GTPase | |||||
| IT-5 | 59 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-5 | 53 | M | + | + | + | c.2729T>A | p.V910D | GE | |||||
| IT-6 | 47 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-6 | 46 | F | + | + | + | c1410_1443 + 4del(38) | p.D470fsX13 | – | |||||
| IT-7 | 42 | M | + | + | + | + | c.1745A>G | p.Y582C | Dynamin | ||||
| NO-1 | 60 | M | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 64 | F | + | + | + | + | + | Spasticity | c.768C>G | p.S256R | CC | ||
| c.854A>G | p.Q285R | GTPase | |||||||||||
| NO-1 | 43 | F | + | c.768C>G | p.S256R | CC | |||||||
| NO-1 | 35 | F | + | c.768C>G | p.S256R | CC | |||||||
| US-1 | 71 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 68 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 56 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 77 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 64 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 49 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 45 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 38 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 42 | M | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 43 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 51 | F | + | + | + | + | c.1334G>A | p.R445H | GTPase | ||||
| US-1 | 26 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-1 | 19 | F | + | + | + | c.1334G>A | p.R445H | GTPase | |||||
| US-2 | 48 | F | + | + | + | c113_130del(18) | p.R38_S43del | – | |||||
AU = Austria; BE = Belgium; BR = Brazil; CN = China; DE = Germany; ES = Spain; FI = Finland; FR = France; IT = Italy; NO = Norway; UK = United Kingdom; US = USA; AA = amino acid; FHx = family history; HSP = hereditary spastic paraplegia; MS = multiple sclerosis-like illness; CAPOS = cerebellar ataxia, areflexia, pes cavus, optic atrophy and sensorineural hearing loss; CC = coiled-coil domain; GE = GTPase effector domain; PEO = progressive external ophthalmoplegia.
aAge of death.
bDe novo OPA1 mutation.
cIn-frame skipping of exon 12 (p.T381-N404del).
BE-1: (Meire et al., 1985; Payne et al., 2004); CN-1: (Li et al., 2005); CN-2: (Ke et al., 2006); CN-3: (Chen et al., 2007); ES-1: (Amati-Bonneau et al., 2005); FI-1: (Puomila et al., 2005); FR-6, FR-7, FR-8:(Amati-Bonneau et al., 2005); FR-9, FR-10: (Amati-Bonneau et al., 2008); IT-4: (Amati-Bonneau et al., 2008; Liguori et al., 2008); IT-5: (Amati-Bonneau et al., 2008); IT-6: (Spinazzi et al., 2008); IT-7: (Ferraris et al., 2008); UK-11: (Hudson et al., 2008); UK-12, UK-13, UK-14: (Stewart et al., 2008); US-1: (Payne et al., 2004); US-2: (Milone et al., 2009).
OPA1 mutations in DOA+
A total of 33 different OPA1 mutations were identified in these 45 independent DOA+ families: missense (n = 18, 54.5%), nonsense (n = 2, 6.1%), splice site (n = 8, 24.2%), and deletion (n = 5, 15.2%) (Table 1, Fig. 1). A majority of these were located in the GTPase regions (n = 19, 57.6%), with a small cluster in the dynamin domain (n = 4, 12.1%). We compared our findings to the eOPA1 database (http://lbbma.univ-angers.fr/lbbma.php?id=9), which currently lists 205 different OPA1 mutations, all of which have been reported in patients with non-syndromal pure DOA. There was a higher risk of developing a DOA+ phenotype with missense mutations (OR = 3.06, 95% CI = 1.44-6.49, P = 0.0027), and mutations located in the GTPase region (OR = 2.29, 95% CI = 1.08–4.82, P = 0.0271) (Table 2). The risk of this occurring was significantly lower with mutations within the dynamin gene domain (OR = 0.28, 95% CI = 0.10–0.84. P = 0.0159) (Table 2).
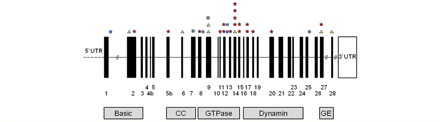
Diagrammatic representation of the OPA1 gene with the location of mutations resulting in DOA+ syndromes. The mutation type is indicated by (i) red stars (missense); (ii) grey squares (nonsense); (iii) blue circles (splice site); and (iv) green triangles (deletion). CC = coiled-coil domain; GE = GTPase effector domain; UTR = untranslated region.
Odd ratios for developing a DOA+ phenotype based upon OPA1 mutation type and domain
| OPA1 mutations | eOPA1a | This studyb | OR | 95% CI | P-value | |
|---|---|---|---|---|---|---|
| Type (n) | Missense | 55 | 18 | 3.06 | 1.44–6.49 | 0.0027* |
| Nonsense | 34 | 2 | 0.31 | 0.07–1.34 | 0.0975 | |
| Splice | 55 | 8 | 0.81 | 0.35–1.92 | 0.6378 | |
| Deletion | 51 | 5 | 0.50 | 0.18–1.38 | 0.1745 | |
| Domain (n) | GTPase | 76 | 19 | 2.29 | 1.08–4.82 | 0.0271* |
| Dynamin | 67 | 4 | 0.28 | 0.10–0.84 | 0.0159* | |
| Others | 61 | 10 | 1.02 | 0.46–2.27 | 0.9628 |
| OPA1 mutations | eOPA1a | This studyb | OR | 95% CI | P-value | |
|---|---|---|---|---|---|---|
| Type (n) | Missense | 55 | 18 | 3.06 | 1.44–6.49 | 0.0027* |
| Nonsense | 34 | 2 | 0.31 | 0.07–1.34 | 0.0975 | |
| Splice | 55 | 8 | 0.81 | 0.35–1.92 | 0.6378 | |
| Deletion | 51 | 5 | 0.50 | 0.18–1.38 | 0.1745 | |
| Domain (n) | GTPase | 76 | 19 | 2.29 | 1.08–4.82 | 0.0271* |
| Dynamin | 67 | 4 | 0.28 | 0.10–0.84 | 0.0159* | |
| Others | 61 | 10 | 1.02 | 0.46–2.27 | 0.9628 |
CI = confidence interval, OR = odds ratio.
aeOPA1 online database (http://lbbma.univ-angers.fr/lbbma.php?id=9).
bMeta-analysis of 33 OPA1 mutations which includes nine previously published OPA1 mutations. * = Significant P-value.
Odd ratios for developing a DOA+ phenotype based upon OPA1 mutation type and domain
| OPA1 mutations | eOPA1a | This studyb | OR | 95% CI | P-value | |
|---|---|---|---|---|---|---|
| Type (n) | Missense | 55 | 18 | 3.06 | 1.44–6.49 | 0.0027* |
| Nonsense | 34 | 2 | 0.31 | 0.07–1.34 | 0.0975 | |
| Splice | 55 | 8 | 0.81 | 0.35–1.92 | 0.6378 | |
| Deletion | 51 | 5 | 0.50 | 0.18–1.38 | 0.1745 | |
| Domain (n) | GTPase | 76 | 19 | 2.29 | 1.08–4.82 | 0.0271* |
| Dynamin | 67 | 4 | 0.28 | 0.10–0.84 | 0.0159* | |
| Others | 61 | 10 | 1.02 | 0.46–2.27 | 0.9628 |
| OPA1 mutations | eOPA1a | This studyb | OR | 95% CI | P-value | |
|---|---|---|---|---|---|---|
| Type (n) | Missense | 55 | 18 | 3.06 | 1.44–6.49 | 0.0027* |
| Nonsense | 34 | 2 | 0.31 | 0.07–1.34 | 0.0975 | |
| Splice | 55 | 8 | 0.81 | 0.35–1.92 | 0.6378 | |
| Deletion | 51 | 5 | 0.50 | 0.18–1.38 | 0.1745 | |
| Domain (n) | GTPase | 76 | 19 | 2.29 | 1.08–4.82 | 0.0271* |
| Dynamin | 67 | 4 | 0.28 | 0.10–0.84 | 0.0159* | |
| Others | 61 | 10 | 1.02 | 0.46–2.27 | 0.9628 |
CI = confidence interval, OR = odds ratio.
aeOPA1 online database (http://lbbma.univ-angers.fr/lbbma.php?id=9).
bMeta-analysis of 33 OPA1 mutations which includes nine previously published OPA1 mutations. * = Significant P-value.
We identified 9 novel OPA1 variants which were not found in 150–300 age- and ethnically-matched control subjects: (i) c.344C > T (p.A115V) in exon 2; (ii) c.768C > G (p.S256R) in exon 5b; (iii) c.728T > A (p.L243X) in exon 7; (iv) c.854A > G (p.Q285R) in exon 8; (v) c.1071A > G (p.A358A) in exon 11; (vi) c.1327G > A (p.A443T) in exon 14; (vii) c.1462G > A (p.G488R) in exon 15; (viii) c.1484C > T (p.A495V) in exon 15; and (ix) c.1642G > A (p.V548I) in exon 17 (Supplementary Fig. 1). With complementary DNA analysis, in-frame skipping of exon 12, with the loss of 24 amino acids (p.T381-N404del), was demonstrated for the c.1071A > G (p.A358A) synonymous substitution. In two German siblings (DE-1), both the c.635_636del (amino acid) deletion (p.K212fsX4) in exon 6 and the c.1642G > A (p.V548I) missense mutation in exon 17 were present. In the absence of additional clinical information and DNA samples from other family members, it was not possible to determine whether these variants were present on the same allele or were compound heterozygous. A 60-year-old Norwegian proband (NO-1) was compound heterozygous for the c.768C > G (p.S256R) missense mutation in exon 5b and the c.854A > G (p.Q285R) missense mutation in exon 8 (Table 1), and these two variants were also identified in his 64-year-old, similarly affected, sister. Both their parents, who are no longer alive, were apparently healthy, and the proband’s two daughters, who are not visually symptomatic, only harboured the c.768C > G substitution in exon 5b. Haplotype analysis confirmed that the proband and his affected sister were compound heterozygous for the c.768C > G (p.S256R) and the c.854A > G (p.Q285R) mutations.
Major clinical features observed in DOA+ patients
| Clinical featuresa | n | % | 95% CI | |
|---|---|---|---|---|
| Lower | Upper | |||
| Optic atrophy | 89/104 | 85.6 | 77.5 | 91.2 |
| Deafness | 65/104 | 62.5 | 52.9 | 71.2 |
| Ataxia | 31/104 | 29.8 | 21.8 | 39.2 |
| Neuropathy | 31/104 | 29.8 | 21.8 | 39.2 |
| Myopathy | 37/104 | 35.6 | 27.0 | 45.2 |
| Progressive external ophthalmoplegia | 48/104 | 46.2 | 36.9 | 55.7 |
| Clinical featuresa | n | % | 95% CI | |
|---|---|---|---|---|
| Lower | Upper | |||
| Optic atrophy | 89/104 | 85.6 | 77.5 | 91.2 |
| Deafness | 65/104 | 62.5 | 52.9 | 71.2 |
| Ataxia | 31/104 | 29.8 | 21.8 | 39.2 |
| Neuropathy | 31/104 | 29.8 | 21.8 | 39.2 |
| Myopathy | 37/104 | 35.6 | 27.0 | 45.2 |
| Progressive external ophthalmoplegia | 48/104 | 46.2 | 36.9 | 55.7 |
CI = confidence interval.
aMeta-analysis of 104 OPA1 mutation carriers from 45 DOA+ families, which includes previously published data on 44 individuals from 18 DOA+ families.
Major clinical features observed in DOA+ patients
| Clinical featuresa | n | % | 95% CI | |
|---|---|---|---|---|
| Lower | Upper | |||
| Optic atrophy | 89/104 | 85.6 | 77.5 | 91.2 |
| Deafness | 65/104 | 62.5 | 52.9 | 71.2 |
| Ataxia | 31/104 | 29.8 | 21.8 | 39.2 |
| Neuropathy | 31/104 | 29.8 | 21.8 | 39.2 |
| Myopathy | 37/104 | 35.6 | 27.0 | 45.2 |
| Progressive external ophthalmoplegia | 48/104 | 46.2 | 36.9 | 55.7 |
| Clinical featuresa | n | % | 95% CI | |
|---|---|---|---|---|
| Lower | Upper | |||
| Optic atrophy | 89/104 | 85.6 | 77.5 | 91.2 |
| Deafness | 65/104 | 62.5 | 52.9 | 71.2 |
| Ataxia | 31/104 | 29.8 | 21.8 | 39.2 |
| Neuropathy | 31/104 | 29.8 | 21.8 | 39.2 |
| Myopathy | 37/104 | 35.6 | 27.0 | 45.2 |
| Progressive external ophthalmoplegia | 48/104 | 46.2 | 36.9 | 55.7 |
CI = confidence interval.
aMeta-analysis of 104 OPA1 mutation carriers from 45 DOA+ families, which includes previously published data on 44 individuals from 18 DOA+ families.
Major clinical features
Optic nerve dysfunction was present in 89/104 (85.6%) patients with DOA+ features in our cohort (Table 3). In two, large, three-generational families with autosomal dominant progressive external ophthalmoplegia (IT-1 and IT-3), the penetrance of visual failure was low (2/13, 15.4%), and if these are excluded, only four of the remaining 91 cases (4.4%) were visually asymptomatic with normal optic nerve parameters. Visual acuity data were available for 66 patients and they were compared with 42 patients from the same families, but with only isolated optic nerve involvement. The mean age of onset of visual loss was 8.2 years (SD = 0.9 years) for the pure DOA group, and 7.7 years (SD = 0.9 years) for the DOA+ group. This difference was not statistically significant (P = 0.7255) and the majority of individuals in both groups (∼95%) were symptomatic by the age of 15 years (Fig. 2A). At the time of their last ophthalmological assessment, the mean visual acuity was 0.87 LogMAR (6/44 Snellen) for patients with pure DOA, and 1.07 LogMAR (6/70 Snellen) for those with DOA+ phenotypes, this difference being statistically significant (P = 0.0170) (Fig. 2B).
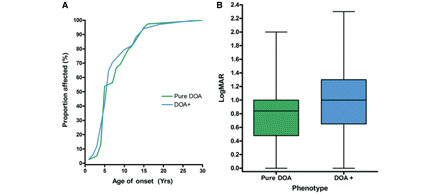
(A) Cumulative distribution curve of age on onset of visual failure in pure DOA and DOA+, and (B) comparison of mean LogMAR visual acuity between patients with pure DOA and DOA+ features (P = 0.0170). The whiskers represent the minimum and maximum LogMAR values, the ends of the boxes are the upper and lower quartiles, the vertical length of the boxes indicate the interquartile range, and the line within the boxes represent the median LogMAR values for each group.
The second most common feature documented in DOA+ patients was deafness (65/104, 62.5%) which invariably followed the onset of visual failure with a peak age of onset in the second and third decades of life (Table 3). Pure-tone threshold audiometry was performed in 58 out of 65 cases and confirmed sensorineural hearing impairment. Of those individuals who suffered from hearing loss, 39 out of 65 (60.0%) also developed significant neuromuscular deficits in later life, and these were a combination of progressive external ophthalmoplegia (48/104, 46.2%), proximal myopathy (37/104, 35.6%), ataxia (31/104, 29.8%), and axonal sensory and/or motor neuropathy (31/104, 29.8%). These additional features became manifest from the third decade of life onwards, with on average a slightly later age of onset for progressive external ophthalmoplegia. Subclinical nerve conduction abnormalities were detected in two of the 31 individuals who were investigated further because of their strong familial phenotype, and neurophysiological studies were normal in nine patients with symptoms suggestive of a possible neuropathic component.
Novel presentations
Spastic paraplegia
In two families with clinically presumed autosomal dominant hereditary spastic paraplegia (HSP), pathogenic OPA1 mutations were identified segregating with both optic atrophy and spastic paraparesis: (i) a 3-bp in-frame deletion c.876-878del(TGT) in exon 9 (p.V294fsX667) (UK-3); and (ii) a nonsense GTPase mutation c.889C > T in exon 9 (p.Q297X) (UK-5), which are predicted to result in premature termination codons. Prior to OPA1 screening, the probands were extensively investigated to establish the underlying cause for their spasticity, and these were all negative: cerebrospinal fluid (CSF) studies, metabolic screen, brain and spinal cord MRI, and SPG4 genetic testing. In family UK-3, the proband was a 59-year-old man who first presented to a neurologist when he was 46-years-old with marked lower limb spasticity, in addition to complicated migraine and poor vision (counting fingers, both eyes), which was noted at the time of preschool vision screening. His spastic paraplegia has gradually worsened over the years and he had more recently developed an axonal sensorimotor neuropathy. His maternal uncle who died at the age of 75-years-old was similarly affected but both of the proband’s two sons have isolated optic atrophy and no subjective problems with their legs. In family UK-5, the proband was a 48-year-old woman with progressive gait and balance difficulties going back to her mid-thirties and severe visual failure from early childhood (counting fingers, both eyes). Her neuro-ophthalmological investigations confirmed bilateral total optic atrophy, lower limb spasticity with spontaneous ankle clonus, and a predominantly axonal sensory neuropathy on nerve conductions studies. Her 21-year-old son was a mutational carrier and although visually asymptomatic (6/6, both eyes), he had subtle temporal disc pallor and lower limb spasticity which had previously been ascribed to cerebral palsy. His 26-year-old sister also had evidence of mild optic nerve involvement (6/9, both eyes) but her neurological examination was otherwise normal.
Multiple sclerosis-like illness
A 58-year-old woman was referred by her optometrist following a routine eye test with suspected bilateral optic atrophy (UK-6). She had no visual complaints (6/9, both eyes) but on examination her colour vision was severely reduced, with bilateral small caeco-central scotomas on Goldmann field perimetry. She had mild gait ataxia and a comprehensive neurological assessment revealed periventricular white matter lesions on MRI, and unmatched oligoclonal bands in her CSF, both supportive of an underlying demyelinating process clinically indistinguishable from multiple sclerosis (Fig. 3). Two of her brothers, aged 61 and 63 years old, had previously been investigated for poor vision and a label of Leber hereditary optic neuropathy was given, even though a screen for the three primary Leber hereditary optic neuropathy mtDNA point mutations (m.3460G > A, m.11778G > A and m.14484T > C) was negative. The family history was also consistent with an autosomal dominant pattern of inheritance and this prompted OPA1 genetic screening, which uncovered a c.2613 + 1g>a splice-site mutation within intron 25 segregating with affected disease status in both the proband and her two brothers.
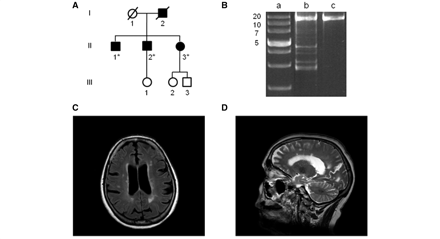
Molecular investigations and MRI neuroimaging of a 58-year-old female presenting with a multiple sclerosis-like illness and harbouring a c.2613 + 1g>a splice-site mutation within intron 25 of the OPA1 gene (UK-6). (A) Family tree of the affected proband (II-3). DNA samples were obtained from her 62-year-old (II-1) and 59-year-old (II-2) brothers, who both had a pure DOA phenotype, and OPA1 sequencing confirmed the presence of the same heterozygous c.2613 + 1g>a variant. The asterisk indicates family members where DNA samples were available for molecular analysis. (B) A 16 kb long-range PCR assay was performed on homogenate DNA extracted from the patient’s skeletal muscle biopsy, and multiple deletion bands were identified. Lane a: 1 kb DNA ladder with the adjacent numbers indicating the size of the band; lane b: patient; lane c: wild-type control. (C) Axial T2-weighted fluid attenuated inversion recovery and (D) sagittal T2-weighted turbo spin echo slices showing cortical atrophy and characteristic white matter lesions in the periventricular areas and more peripherally near to grey matter.
Additional clinical features
Late-onset non-insulin dependent diabetes developed in 3 of the 83 (3.6%) DOA+ patients and this was associated with deafness in one case. Recurrent migrainous-type headaches were also reported by 4 of the 83 (4.8%) patients in our DOA+ cohort: (i) a 36-year-old woman with the c.870+5g > a mutation (UK-2); (ii) her 54-year-old sister who also carried the c.870 + 5g>a mutation; (iii) a 59-year-old man with the c.876_878del(TGT) deletion (UK-3); and (iv) a 38-year-old woman with the c.1635C > G mutation (UK-11). The onset was in their mid to late teens, three of the four (75.0%) described a prodromal phase characterized by positive visual phenomena and photophobia, and all four of them experienced a stereotypical pattern characterized by severe fronto-temporal headaches associated with nausea and vomiting. Additional complicated features included dysphasia, evolving visual scotomas, and a transient confusional state after the headache had subsided.
Neuroimaging and metabolic findings
Neuroimaging abnormalities were identified in 12 of the 40 (30.0%) DOA+ patients: (i) bilateral basal ganglia calcifications (n = 2, 5.0%); (ii) periventricular multiple sclerosis-like lesions (n = 2, 5.0%); (iii) non-specific white matter lesions (n = 4, 10.0%); and (iv) varying degrees of cortical and cerebellar atrophy (n = 5, 12.5%). Brain magnetic resonance spectroscopy was performed on a 43-year-old man with a complex DOA+ phenotype due to a c.1635C > G mutation in exon 17 (FR-9). His MRI scan showed mild cortical and cerebellar atrophy, but both n-acetyl-aspartate and lactate peaks were not altered on his magnetic resonance spectroscopy spectra. CSF analysis performed on 29 DOA+ patients was unremarkable, except for the two patients with periventricular white matter lesions (UK-6 and FR-11), who both had unmatched oligoclonal bands. There was no evidence of marked metabolic derangement in our patient cohort. Plasma lactate levels were elevated in only five patients, and no abnormal organic acids profile was reported in the subgroup of patients with ataxia and spastic paraplegia.
Histochemical studies
Dual COX–SDH staining was performed on skeletal muscle biopsies from 10 patients with pure DOA (mean age = 49.3 years, SD = 9.6 years, range = 39.0-65.0 years) and 24 patients with DOA+ phenotypes (mean age = 50.6 years, SD = 11.0 years, range = 24.0-69.0 years), and there was no significant age difference between these two groups (P = 0.7354). Published muscle biopsy data from an additional four patients with features of DOA+ were also included in the meta-analysis (Ferraris et al., 2008; Spinazzi et al., 2008; Milone et al., 2009). The frequency of COX-deficient muscle fibres in the pure DOA group (mean = 1.30%, SD = 1.20%, range = 0–3.00%) was significantly lower than in the DOA+ group (mean = 5.41%, SD = 5.90%, range = 0–21.18%, P = 0.0226, Fig. 4A). However, COX-deficient fibres were significantly more frequent in both pure DOA (P < 0.0001) and DOA+ patients (P < 0.0001), compared to controls (mean = 0.03%, SD = 0.07%, range = 0–0.30%, Fig. 4B).
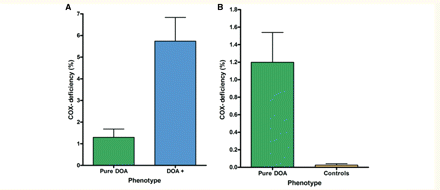
Mean level of COX-deficiency in skeletal muscle biopsies: (A) pure DOA versus DOA+ phenotypes (P = 0.0226) and (B) pure DOA versus age-matched controls (P < 0.0001). The error bars represent the standard error of the mean.
Mitochondrial DNA analysis
There was clear evidence of multiple mtDNA deletions in skeletal muscle in the majority of patients with both pure DOA (8/10, 80.0%, Fig. 5 ) and DOA+ phenotypes (24/28, 85.7%), with no significant difference between these 2 groups (χ2 = 0.1810, P = 0.6706, Table 4). For the four patients with DOA+ features but no multiple mtDNA deletions on long-range PCR, COX-negative fibres were also not detected in their muscle biopsies (mean age = 45.5 years, SD = 7.9 years, range = 35.0–54.0 years). This specific subgroup harboured two different splice-site mutations: c.1212 + 3a>t (UK-1) and c.870 + 5g>a (UK-2), and a previously reported frame-shift deletion c1410_1443 + 4del(38) (Spinazzi et al.2008). Repeat muscle biopsies were available for two patients. The first was a 36-year-old woman with proximal myopathy, peripheral neuropathy, complicated migraine and the c.870 + 5g>a splice-site mutation (UK-2). Muscle biopsies were taken when she was 25- and 35-years-old, and both showed no evidence of COX-deficiency or multiple mtDNA deletions on long-range PCR. In contrast, her 55-year-old sister, who had a remarkably similar clinical picture, had definite evidence of multiple mtDNA deletions on long-range PCR with normal COX-SDH staining in a muscle biopsy taken at the age of 54 years. The second was a 59-year-old man with an HSP-like phenotype, complicated migraine and the c.876_878del(TGT) deletion (UK-3). His most recent muscle biopsy taken when he was 58-years-old revealed 2.07% (10/482) COX-deficient fibres and a laddering pattern on long range PCR indicating multiple deleted mtDNA species. Histochemical and molecular studies on a skeletal muscle specimen taken 13 years earlier, when his lower limb spasticity was much less severe, were entirely normal. There was no evidence of mtDNA depletion in skeletal muscle homogenate DNA from OPA1 positive patients (mean mtDNA copy number = 5144, SD = 2843, n = 24) when compared to age-matched healthy controls (mean mtDNA copy number = 4669, SD = 2640, n = 20, P = 0.5719; Fig. 6).
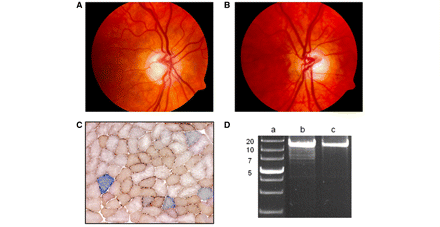
Clinical, histochemical and molecular features observed in a 59-year-old female with isolated optic nerve involvement due to an exon 27 c.2713C>T (p.R905X) OPA1 missense mutation. (A) Right, and (B) left optic discs displaying generalized pallor of the neuro-retinal rim and pathological excavation. (C) Dual COX-SDH histochemistry performed on 20 µm thick cryostat sections showing the presence of COX-deficient muscle fibres (33/1100, 3.00%). (D) Long-range PCR of the patient’s homogenate skeletal muscle DNA revealing multiple deletion bands in addition to the wild-type 16-kb fragment. Lane a: 1-kb DNA ladder with the adjacent numbers indicating the size of the band; lane b: patient; lane c: wild-type control.
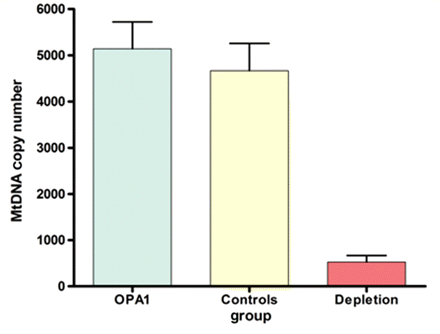
Comparison of mtDNA copy number in homogenate skeletal muscle DNA from patients with OPA1 mutations (mean = 5144, SD = 2843, n = 24) with age-matched healthy controls (mean = 4669, SD = 2640, n = 20, P = 0.5719), and patients with mtDNA depletion syndromes (mean = 524, SD = 345, n = 6, P = 0.0005). The error bars represent the standard error of the mean.
Summary of OPA1 mutations and molecular profile identified in patients with DOA+ phenotypes
| OPA1 mutations | |||||
|---|---|---|---|---|---|
| Missense | Nonsense | Splice site | Deletion | ||
| DOA + patients | n | 80 | 3 | 8 | 13 |
| COX-deficiencya | − | 1 | – | 3 | 2 |
| + | 17 | 1 | 2 | 2 | |
| MtDNA deletionsa | − | – | – | 2 | 2 |
| + | 18 | 1 | 3 | 2 | |
| OPA1 mutations | |||||
|---|---|---|---|---|---|
| Missense | Nonsense | Splice site | Deletion | ||
| DOA + patients | n | 80 | 3 | 8 | 13 |
| COX-deficiencya | − | 1 | – | 3 | 2 |
| + | 17 | 1 | 2 | 2 | |
| MtDNA deletionsa | − | – | – | 2 | 2 |
| + | 18 | 1 | 3 | 2 | |
aFor DOA+ patients where muscle biopsies were available for histochemical and molecular analysis (n = 28).
Summary of OPA1 mutations and molecular profile identified in patients with DOA+ phenotypes
| OPA1 mutations | |||||
|---|---|---|---|---|---|
| Missense | Nonsense | Splice site | Deletion | ||
| DOA + patients | n | 80 | 3 | 8 | 13 |
| COX-deficiencya | − | 1 | – | 3 | 2 |
| + | 17 | 1 | 2 | 2 | |
| MtDNA deletionsa | − | – | – | 2 | 2 |
| + | 18 | 1 | 3 | 2 | |
| OPA1 mutations | |||||
|---|---|---|---|---|---|
| Missense | Nonsense | Splice site | Deletion | ||
| DOA + patients | n | 80 | 3 | 8 | 13 |
| COX-deficiencya | − | 1 | – | 3 | 2 |
| + | 17 | 1 | 2 | 2 | |
| MtDNA deletionsa | − | – | – | 2 | 2 |
| + | 18 | 1 | 3 | 2 | |
aFor DOA+ patients where muscle biopsies were available for histochemical and molecular analysis (n = 28).
Discussion
By studying 45 DOA+ families from different geographical backgrounds, we have shown that additional neuromuscular features frequently develop as the disease progresses, with an adverse impact on the pre-existing visual morbidity. A meta-analysis of 104 patients with DOA+ phenotypes, including 44 previously reported cases in the literature, confirmed that sensorineural deafness is the most frequently observed extra-ocular feature, present in nearly two-thirds of all patients, over half of whom eventually developed more severe neuromuscular symptoms. Progressive external ophthalmoplegia and a predominantly axonal sensory neuropathy were the next most common clinical manifestations, followed by ataxia and myopathy. Although there is a degree of overlap, a chronological pattern is emerging with visual failure occurring within the first decade of life, followed by deafness from late childhood to early adulthood, and then a combination of ataxia, myopathy, peripheral neuropathy and progressive external ophthalmoplegia from the third decade of life onwards (Fig. 7). Excluding singleton cases and subjects younger than 25-years-old, it is also remarkable that in about a third of all pedigrees (11/32, 34.4%), family members were found to manifest both pure DOA and DOA+ phenotypes despite carrying the same OPA1 mutation, and presumably having shared common environmental influences, especially in the first two decades of life in parent-offspring cases.
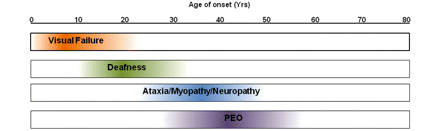
Evolution of the major clinical features observed in DOA+ syndromes.
Optic nerve sparing
OPA1 mutations were identified in two large Italian pedigrees (IT-1 and IT-3) segregating autosomal dominant progressive external ophthalmoplegia, and remarkably, only a minority of family members were visually symptomatic with evidence of optic atrophy (2/13, 15.4%). In sharp contrast, almost all of the remaining DOA+ cases had evidence of optic nerve dysfunction (87/91, 95.6%), and the published penetrance figures are generally >80% in well characterized, multi-generational families with pure DOA (Yu-Wai-Man et al., 2009a). The observation in these two Italian families has important diagnostic implications, and it will be important to screen a larger cohort of autosomal dominant progressive external ophthalmoplegia pedigrees without prominent visual symptoms, to determine the true prevalence of OPA1 mutations in this specific disease subgroup. Furthermore, it is intriguing that OPA1 mutational carriers can be non-penetrant for optic nerve failure, but yet manifest other deficits including deafness, myopathy and peripheral neuropathy, besides progressive external ophthalmoplegia. This is critically important, because it means that clinicians should consider OPA1 screening in patients with unexplained neurological features, especially if molecular investigations subsequently reveal the presence of multiple mtDNA deletions (Stewart et al., 2008).
Expanding DOA+ phenotype
We indentified several unexpected phenotypes in patients with OPA1 mutations, including spastic paraparesis and a multiple sclerosis-like illness, and these are unlikely to be chance associations. In the two probands presenting with progressive lower limb spasticity (UK-3 and UK-5), no underlying metabolic, inflammatory or compressive causes were found despite extensive investigations over several years. Isolated spastic paraparesis is unusual in adults with mitochondrial disease (Corona et al., 2002), and the degeneration of corticospinal tract axons seen in these two families is particularly revealing. Further studies into the mechanisms involved could provide important insights into the pathophysiological mechanisms at work in hereditary spastic paraplegia, the obvious corollary being that the OPA1 mutation is having a negative impact on neuronal survival by creating an imbalance in mitochondrial network dynamic and axonal trafficking (Reid, 2003; Salinas et al., 2008). The presence of COX-deficient fibres and multiple mtDNA deletions in skeletal muscle biopsies is also very significant and further supports a link with mitochondrial dysfunction in a subgroup of patients with hereditary spastic paraplegia (McDermott et al., 2003).
The unmatched CSF oligoclonal bands and abnormal white matter lesions in the 58-year-old woman with a multiple sclerosis-like illness (UK-6) are also unlikely to be co-incidental findings. Based upon recent epidemiological data (Fox et al., 2004), we estimate the chance finding of an OPA1 mutation in a patient with multiple sclerosis in the UK at 1 in 41 million (95% CI = 25–73 million), and of note, this association has already been reported in a 44-year-old man with gait ataxia and lower-limb spasticity (FR-11), who had MRI and CSF changes consistent with a demyelinating process, and a novel c.1937C > T (p.S646L) missense mutation in the dynamin region the OPA1 gene (Verny et al., 2008). These observations are reminiscent of the well-established association between primary mtDNA Leber hereditary optic neuropathy mutations and a multiple sclerosis-like illness (Harding’s disease) (Harding et al., 1992; Kovacs et al., 2005), and it is therefore tempting to speculate a causal link between OPA1 mutations, mitochondrial dysfunction, oligodendrocyte survival and the complex autoimmune process which underlies the pathophysiology of multiple sclerosis (Carelli and Bellan, 2008; Mahad et al., 2009).
Nearly a third of all the DOA+ patients in our study had evidence of an axonal neuropathy, an observation with important parallels to a specific subtype of Charcot-Marie-Tooth (CMT) disease or, hereditary motor and sensory neuropathy type VI (HMSN VI, OMIM 601152). This variant is caused by mutations in the MFN2 gene and it is characterized by early onset, severe peripheral neuropathy in the first decade of life, followed by subacute, progressive optic atrophy in later childhood (Zuchner et al., 2006). Mitofusin-2 is located within the mitochondrial outer membrane and it shares a high degree of structural and functional complementarity with Opa1, both being dynamin GTPase proteins (Zuchner et al., 2006; Chan, 2007). Mitofusin-2 interacts closely with Opa1 to promote fusion of the mitochondrial network, and it could also play a distinct role in mitochondrial oxidative output by regulating the expression of nuclear-encoded respiratory chain subunits (Pich et al., 2005). It is also noteworthy that abnormal nerve conduction studies were documented in two DOA+ patients with no overt neuropathic features. As neurophysiological investigations were performed mostly to confirm a suspected peripheral neuropathy, the true burden of subclinical disease remains to be determined, not only in DOA+, but also among patients with pure DOA.
The prevalence of non-insulin dependent diabetes in our DOA+ cohort (3.6%) was comparable to the latest UK national figures (3.9%) (NHS Quality and Outcomes Framework, 2008), and although severe, characteristic migrainous episodes were reported by 4 out of 83 (4.8%) patients with DOA+, this could simply reflect the background prevalence of migraine in the general population (11–15%), and not an OPA1-related effect per se (Goadsby et al., 2002; Bigal et al., 2006).
Prevalence of DOA+
As a result of the different referral patterns in this study, the prevalence of patients with DOA+ phenotypes is difficult to ascertain. However, a reasonable estimate can be derived from our epidemiological data from the North of England, which has a centralized National Health Service with well defined catchment areas (Man et al., 2003). Based upon our latest figures, 64 patients with OPA1 mutations currently reside in the North of England, and of those 11 out of 64 (17.2%) had DOA+ features, suggesting that these syndromal manifestations are not uncommon and probably affect up to one in six of all cases. Using the latest census data for the North of England (Office for National Statistics, 2008), we estimate the minimum prevalence of DOA+ at 0.36 per 100 000 (95% CI = 0.25–0.50 per 100 000); about 1 in 250 000 of the general population. This is comparable to other well-recognized neurogenetic disorders with prominent ocular features such as oculopharyngeal muscular dystrophy (Brais, 2009), neuromyelitis optica (Devic disease) (Wingerchuk et al., 2007), and diabetes insipidus and mellitus with optic atrophy and deafness (Wolfram syndrome 1) (Khanim et al., 2001).
Novel OPA1 mutations
We have identified an additional nine novel OPA1 mutations in this study, adding to the list of >200 pathogenic variants so far catalogued on the eOPA1 database (Ferre et al., 2005, 2009). Unusually, in two families, DE-1 and NO-1, affected family members harboured two different OPA1 mutations. Only one other case of compound heterozygosity has previously been described in a female patient from a German family, and she carried two presumed pathogenic substitutions in exon 8: c.808G > A (p.E270K) and c.868C > T (p.R290W) (Pesch et al., 2001). The patient’s visual loss was more severe compared with five other affected family members who were all simple heterozygotes, which led the authors to speculate that her greater visual impairment was related to the presence of two pathogenic OPA1 variants. Unlike this previously published family who only exhibited isolated optic atrophy, both DE-1 and NO-1 had DOA + features, with the two Norwegian siblings having a particularly aggressive disease course characterized by ataxia, peripheral neuropathy, corticospinal tract involvement and myopathy, in contrast to relatively mild sensorineural hearing loss for the two German siblings. Although the number of cases is small, it is tempting to invoke a combined mutational effect, with the presence of two OPA1 mutations leading to greater disease severity.
The Norwegian family, NO-1, is also unusual in carrying an exon 5b missense mutation, c.768C > G (p.S256R), in the coiled-coil domain of the Opa1 protein. Using in silico modelling, the change of a serine to arginine at position 256 is predicted to result in a marked reduction in the likelihood of coiled-coil formation in this region (Fig. 8), with a detrimental effect on the ability of the Opa1 protein to form oligomers, which is critical for its normal function. We previously reported another exon 5b mutation, c.740G > A (p.R247H), in a 23-year-old male with pure DOA and a unique clinical course characterized by spontaneous, progressive visual recovery (Cornille et al., 2008). This amino acid substitution was also identified in the patient’s father who was non-penetrant for optic atrophy. The c.740G > A (p.R247H) mutation was predicted to disrupt the coiled-coil forming potential, and in addition, fibroblasts harvested from both family members showed mitochondrial network abnormalities and oxidative phosphorylation uncoupling, with a greater susceptibility to undergo apoptosis (Cornille et al., 2008).
In silico prediction of coiled-coil regions within the segment of 37 amino acids encoded by exon 5b: (A) wild-type, and (B) p.S256R substitution. The modelling accuracy increases with larger scanning windows (28 > 21 > 14), and probabilities > 0.5 indicate a high likelihood of coiled-coil conformation. The p.S256R substitution is predicted to reduce the coiled-coil forming potential by ∼45%. (http://www.ch.embnet.org/software/COILS_form.html). Green line = window 14; Blue line = window 21; Red line = window 28.
It is therefore interesting that only the c.768C > G (p.S256R) variant was identified in the two, visually asymptomatic, 35- and 43-year-old daughters of the Norwegian proband. OPA1 mRNA exists as eight isoforms as a result of alternate splicing of exons 4, 4b and 5b, and from an evolutionary perspective, exons 4b and 5b are only found in higher vertebrates with a putative role in regulating cytochrome c release during apoptosis (Olichon et al., 2007). The 37 amino acids encoded by exon 5b are identical in all higher vertebrates and only two substitutions have so far been reported, both non-synonymous. Although speculative, as exon 5b is only present in ∼40% of total OPA1 mRNA transcripts, this could explain the relatively mild pathogenic nature of exon 5b variants on their own. Additional functional studies, for example comparing fibroblast cell lines from NO-1 family members, are also warranted to further clarify whether there is a synergistic action between the exon 5b and the exon 8 variants in compound heterozygous individuals, contributing to disease severity.
Mitochondrial mechanisms in DOA+
In our original description of multiple mtDNA deletions in DOA+, all the OPA1 mutations were missense substitutions and in 5/7 families, these clustered in the GTPase domain (Amati-Bonneau et al., 2008; Hudson et al., 2008). A possible dominant negative effect was therefore suggested with the mutant Opa1 protein disrupting mitochondrial inner membrane function and/or the delicate balance of the intra-mitochondrial nucleotide pool required for efficient mtDNA replication (Zeviani, 2008). Although these remain to be proven experimentally, a gain of function mechanism cannot explain the association of OPA1 deletions and splice-site mutations with DOA+, both of which lead to haploinsufficiency, and were identified in DOA+ families in this study.
Histochemical and molecular studies performed on skeletal muscle biopsies from patients with both pure DOA and DOA+ features have also provided some interesting insights into the possible disease mechanisms involved in retinal ganglion cell loss and optic nerve degeneration. COX-deficiency is a useful surrogate marker of the extent of mitochondrial dysfunction due to mtDNA defects, and consistent with this, patients with DOA+ phenotypes had over four times higher levels of COX-negative muscle fibres compared to patients with pure DOA. The majority of patients in both subgroups harboured multiple mtDNA deletions, but in a few patients with DOA+ features, both COX-deficiency and secondary mtDNA deletions were not present, even when using long-range PCR, a highly sensitive technique that preferentially amplifies smaller mtDNA species (Lightowlers et al., 1999). It is therefore possible that mtDNA deletions contribute to the pathophysiology of DOA, but they are not the sole mechanism responsible for triggering cellular dysfunction, especially optic nerve neurodegeneration. In keeping with this hypothesis, we recently showed that secondary mtDNA abnormalities were not present in retinal ganglion cells harvested from a pure DOA mouse model, implying that retinal ganglion cell loss must be the result of other deleterious consequences induced by the OPA1 mutation (Yu-Wai-Man et al., 2009b). One important caveat is that the molecular observations in this study relate to skeletal muscle biopsies, which might not be a true reflection of the changes operating within retinal ganglion cells, tissue-specific mechanisms being well-recognized features of mitochondrial disorders.
The Opa1 protein is involved in several inter-related biological processes and further studies are needed to clarify whether specific mutations operate predominantly via certain pathways (Davies and Votruba, 2006; Yu-Wai-Man et al., 2009a). These are not however mutually exclusive, for example, increased levels of reactive oxygen species secondary to disturbed oxidative phosphorylation could potentiate the formation of mtDNA deletions (Krishnan et al., 2008), and mitochondrial network fragmentation could accelerate their clonal expansion, the compartmentalization of the mtDNA pool effectively decreasing the copy number in these discrete units, and reducing the time required for the deleted mtDNA species to reach supra-threshold levels (Chinnery and Samuels, 1999).
Genotype–phenotype correlations
Genetic counselling has always been a challenging issue in DOA due to the marked intra- and inter-familial variability in visual prognosis, and these difficulties have now been further compounded by the heterogeneous individual manifestations of DOA+ features. Although further studies are warranted, our study has allowed us to define some broad genotype–phenotype correlations for clinicians advising patients on the implications of the OPA1 mutations segregating in their families: (i) mutations causing optic atrophy in association with sensorineural deafness are not limited to the c.1334G > A (p.R445H) variant but, as in other reports, we found this mutation to be the most common, being present in 11 out of 30 (36.7%) families in our study; (ii) there is a 2–3-fold increased risk of developing DOA+ features with missense mutations and those located within the GTPase region; (iii) this risk is 4-fold lower with OPA1 mutations located within the dynamin domain; (iv) patients with DOA+ phenotypes have on average a worse visual prognosis compared to patients with pure DOA; and (v) OPA1 is a highly polymorphic gene but there is no evidence that certain combination of single nucleotide polymorphisms are acting as intragenic modifiers and influencing either disease severity or specific clinical manifestations (data not shown).
Conclusions
Although the majority of patients with OPA1 disease have symptomatic optic neuropathy, additional neuromuscular features are common in patients with OPA1 mutations. There is currently no definitive treatment for this disorder, but the fact that these complex phenotypes eventually develop in up to 20% of all mutational carriers has important implications for clinical management. Significant neurological impairment can easily be overlooked in patients and simply attributed to their visual failure, but careful surveillance of at-risk individuals will identify components which might be amenable to supportive therapy, such as physiotherapy or anti-spasmodic drugs for those with upper motor neuron involvement, and cochlear implantation which is of proven benefit in mitochondrial hearing impairment (Sinnathuray et al., 2003; Chinnery and Griffiths, 2006). The heterogeneous multi-system manifestations seen in DOA+ families and the remarkable variable penetrance among carriers with the same pathogenic variant clearly implicate the influence of secondary modulatory factors on the phenotypic expression of the ‘primary’ OPA1 mutation. Identifying these together with the underlying molecular pathways will be key in dissecting the complex processes leading to tissue-specific dysfunction in both pure and syndromal forms of DOA.
Funding
MRC Clinical Research Fellowship in Neuro-ophthalmology to P.Y.W.M.; Wellcome Trust Senior Fellowship in Clinical Science to P.F.C.; P.F.C. also receives funding from the Parkinson’s Disease Society (UK), the Medical Research Council Translational Muscle Centre, and the UK NIHR Biomedical Research Centre in Ageing and Age related disease. R.H. is supported by the Deutsche Forschungsgemeinschaft (HO 2505/2-1) and the Academy of Medical Sciences. R.M. and M.V. are MRC Clinician Scientists. D.M.T. and R.W.T. are funded by the Wellcome Trust, and the UK National Commissioning Group for Rare Mitochondrial Disorders of Adults and Children. L.B. receives funding from Helse Vest (RHF) and the Norwegian Research Council. G.S.G. is supported by the NIHR Biomedical research Centre for Ageing and Age-related disease; Telethon-Italy grant (GGP06233) to V.C.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- COX
cytochrome c oxidase
- DOA
autosomal dominant optic atrophy
- DOA+
dominant optic atrophy plus syndrome
- GTP
Guanosine triphosphate
- LogMAR
logarithm of the minimum angle of resolution
- mtDNA
mitochondrial DNA
- PCR
polymerase chain reaction
- SDH
succinate dehydrogenase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}