




Abstract
Neuromyelitis optica (NMO) is a severe inflammatory CNS disorder of putative autoimmune aetiology, which predominantly affects the spinal cord and optic nerves. Recently, a highly specific serum reactivity to CNS microvessels, subpia and Virchow–Robin spaces was described in patients with NMO [called NMO–IgG (NMO–immunoglobulin G)]. Subsequently, aquaporin-4 (AQP4), the most abundant water channel in the CNS, was identified as its target antigen. Strong support for a pathogenic role of the antibody would come from studies demonstrating a correlation between AQP4-Ab (AQP4-antibody) titres and the clinical course of disease. In this study, we determined AQP4-Ab serum levels in 96 samples from eight NMO–IgG positive patients (median follow-up 62 months) in a newly developed fluorescence-based immunoprecipitation assay employing recombinant human AQP4. We found that AQP4-Ab serum levels correlate with clinical disease activity, with relapses being preceded by an up to 3-fold increase in AQP4-Ab titres, which was not paralleled by a rise in other serum autoantibodies in one patient. Moreover, AQP4-Ab titres were found to correlate with CD19 cell counts during therapy with rituximab. Treatment with immunosuppressants such as rituximab, azathioprine and cyclophosphamide resulted in a marked reduction in antibody levels and relapse rates. Our results demonstrate a strong relationship between AQP4-Abs and clinical state, and support the hypothesis that these antibodies are involved in the pathogenesis of NMO.
Introduction
Neuromyelitis optica (NMO) is a severe inflammatory central nervous system (CNS) disorder of putative autoimmune aetiology, which predominantly affects the spinal cord and optic nerves (Wingerchuk et al., 1999, 2006; de Seze et al., 2002). Recently, a highly specific serum reactivity to CNS microvessels and subpia was described in patients with NMO [called NMO–immunoglobulin G (NMO–IgG)] (Lennon et al., 2004). Subsequently, the same group identified aquaporin-4 (AQP4), the most abundant water channel in the CNS, as the target antigen (Lennon et al., 2005). Both findings have been confirmed by others (Jarius et al., 2007; Takahashi et al., 2007; Paul et al., 2007). There is increasing evidence that NMO–IgG/AQP4-Ab (antibody) contributes to the pathogenesis of the disease (Lucchinetti et al., 2002; Wingerchuk et al., 2007; Jarius et al., 2008). Sites of intralesional AQP4 loss were histopathologically found to correlate with sites of immunoglobulin and complement activation (Misu et al., 2007; Roemer et al., 2007), and the antibody is predominantly IgG1 subclass and activates complement after binding to extracellular epitopes in vitro (Hinson et al., 2007; Waters et al., 2008). Support for a pathogenic role of the antibody would come from studies demonstrating correlation of AQP4-Ab titres and clinical course.
In the present study, we assessed AQP4-Ab in NMO patients with long-term follow-up using a newly developed immunoprecipitation assay employing enhanced green fluorescent protein (EGFP)-tagged recombinant human AQP4 (Waters et al., 2008).
Patients and Methods
Serum samples from eight NMO–IgG-positive patients of Caucasian origin diagnosed with either isolated longitudinally extensive transverse myelitis (LETM) (n = 2) or LETM and optic neuritis (n = 6) were retrospectively evaluated for AQP4-Abs. NMO–IgG testing was done by the Mayo Medical Laboratories (Lennon et al., 2004). Six patients fulfilled Wingerchuk's revised diagnostic criteria (Wingerchuk et al., 2006); the two patients with remitting LETM are part of the NMO-spectrum, a broader clinical syndrome than originally described (Wingerchuk et al., 2007). No history of disease outside the optic nerve or spinal cord was present at onset. Extra-opticospinal MRI lesions were detectable in two patients at disease onset and in five of eight patients (71%) later in the disease course. Disease followed a relapsing course in all patients. Median follow-up was 62 months (range 33–114). Seven patients were female, one male. Median age at onset was 45 years (range 14–59). Serum samples were stored at −80°C until testing. The clinical course was retrospectively evaluated without knowledge of the AQP4-Ab test results. The study was approved by the institutional review boards of the City of Vienna and the Innsbruck Medical University, and patients’ consent was obtained in all cases.
AQP4-Abs were assessed in a fluorescence based immunoprecipitation assay (FIPA) as described in detail elsewhere (Waters et al., 2008). Briefly, 25 µl of each serum was incubated with 250 µl of an extract from human embryonic kidney cells transfected with EGFP-tagged M1- and M23-human AQP4. The IgG was then precipitated using Protein A sepharose beads, washed thoroughly, and the amount of EGFP–AQP4 bound by antibody detected by counting the green fluorescence [arbitrary fluorescence units (FU)] at 512 nm (excitation 472 nm; cut-off 495 nm) on a fluorescence plate reader (SpectraMAx Gemini XS, Molecular Devices, CA, USA). Results were given as FU precipitated by each serum sample under standard conditions. The mean + 3 SD from 10 healthy control samples was 63 FU. Acetylcholine receptor (AChR) antibodies were detected by a commercially available radioimmunoprecipitation assay using 125I-bungarotoxin (DLD Diagnostika, Hamburg, Germany). Antibodies to thyroid peroxidase (TPO) and thyroglobulin (TG) were detected by two commercially available chemoluminescence immunoassays (Immunlite 2000 system; DPC-Buehlmann, Salzburg, Austria). The proportion of CD19-positive cells among total lymphocytes was established by standard flowcytometric analysis of whole-blood samples using a Cytomic FC 500® cell counter (Beckman Fullerton, CA, USA) and IOTest® CD19 PC7 conjugated antibody (Immunotech S.A., Marseille, France) (Pat. 1, 3 and 4) or a FACScan (BD Biosciences, NJ, USA) and tritest CD45/CD3/CD19 antibodies (BD Biosciences, NJ, USA) (reference range: 0.1–0.5 × 109 cells/l or 6–19% of the total lymphocyte number) (Pat. 2). The protocols for flowcytometric analysis are approved for diagnostic use.
Results
AQP4-Ab was determined in 96 samples (median 10/patient; range 7–18) from eight patients previously found to be NMO–IgG positive. Ninety-five out of 96 samples samples were positive for AQP4-Ab. AQP4-Ab values varied between 61 and 1091 FU (median 302; cut-off 63). Detailed results are shown in Figs 1–3.
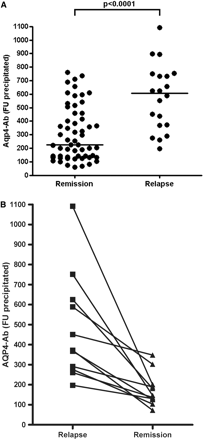
AQP4-Ab levels during relapse and remission. (A) Median AQP4-Ab levels from 57 samples stratified according to disease activity (P < 0.0001; Mann Whitney test). (B) Maximum AQP4-Ab serum levels from 11 samples taken during relapses in six patients and Nadir values during subsequent remission (P = 0.001; Wilcoxon matched-pairs signed-rank test).


AQP4-Ab levels, CD19 cell counts (% of total lymphocytes), relapses and immunosuppressive treatment over time in eight patients with NMO. Time points are selected to illustrate the relationship between AQP4-Ab, relapses and therapies. (A and J) Pat.1; (B) Pat.3; (C) Pat.2; (D and H) Pat. 7; (E) Pat. 8; (F) Pat. 6; (G) Pat. 5; (I) Pat. 4. See results section for details. ♦ = AQP4-Ab serum levels;  = CD19 cell counts;
= CD19 cell counts;  = clinical relapse;
= clinical relapse;  = intravenous methylprednisolone;
= intravenous methylprednisolone;  = prednisolone;
= prednisolone;  = azathioprine;
= azathioprine;  = dexamethasone;
= dexamethasone;  = rituximab;
= rituximab;  = cyclophosphamide;
= cyclophosphamide;  = mitoxantrone;
= mitoxantrone;  = plasma exchange; eod = every other day.
= plasma exchange; eod = every other day.
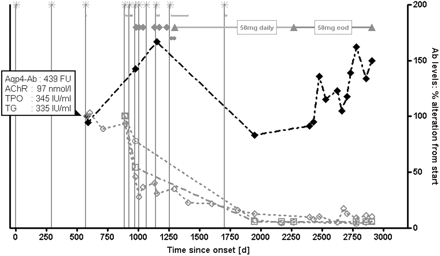
Increase in AQP4-Ab levels during relapse is not paralleled by a rise in other autoimmune autoantibodies. ♦ = AQP4-Ab;  = AChR-Ab;
= AChR-Ab;  = TG-Ab;
= TG-Ab;  = TPO-Ab; = clinical relapse; = intravenous methylprednisolone; = prednisolone; = azathioprine; = cyclophosphamide; = mitoxantrone.
= TPO-Ab; = clinical relapse; = intravenous methylprednisolone; = prednisolone; = azathioprine; = cyclophosphamide; = mitoxantrone.
AQP4-Ab serum levels in relapse and remission
AQP4-Ab was determined in 20 samples from eight patients obtained at onset of relapse. Median AQP4-Ab levels in these samples were higher (607 FU; range 198–1091 FU) when compared with samples taken during remission (median 221; range 61–761; n = 57) (P < 0.0001, Mann Whitney test; Fig. 1A), and maximum AQP4-Ab levels in relapse were significantly higher than nadir values during the following remission period in paired samples (P < 0.001; Wilcoxon's matched-pairs rank sum test; Fig. 1B). The 19/20 (95%) samples taken during relapse yielded results exceeding the overall median AQP4-Ab levels found during remission. In five cases, samples taken within 100 days prior to onset of relapse were available, demonstrating that NMO attacks are preceded by a marked rise in AQP4-Ab levels (Fig. 2A–E). Antibody values rose by 124–294% (median 192) within 48–99 days (median 85), corresponding to a median increase of around 20% per week prior to relapse.
Conversely, AQP4-Ab was detectable in 56/57 samples from eight patients during remission (>30 days from relapse onset), but the titres were low (<2 × cut-off) in eight samples and slightly under cut-off in one (Fig. 2I). The 50/57 remission samples (88%) yielded results below the overall median AQP4-Ab value found during relapse.
Increase in AQP4-Ab levels during relapse is not paralleled by a rise in other autoimmune autoantibodies
In one patient with pre-existing autoimmune myasthenia gravis and autoimmune thyroditis, AQP4-Ab was determined in parallel with antibodies to AChR, TPO and TG. While AQP4-Ab levels increased during two relapses by 42% and 67% compared with the first available value, all other autoantibodies titres had declined at that time (AChR-Ab by 54.1%; TPO-Ab 32%; TG-Ab 45.7%) (Fig. 3). Moreover, AQP4-Ab levels rose by 49.8% over the observation period of 2323 days, while AChR-, TPO- and TG-Ab concentrations declined by 90%, 94.2% and 94.3%, respectively, under therapy with cyclophosphamide. While AChR-, TPO- and TG-Ab titres correlated well over time (r2 = 0.9, 0.95 and 0.99, respectively; P < 0.005), no correlation of AQP4-Ab levels with any of these autoantibodies was found (r2 < 0.04).
Prompt and rapid decline of AQP4-Ab levels under therapy
Twelve follow-up samples from seven patients were obtained within 100 days (median 37; range 6–92) from onset of relapse. AQP4-Ab testing demonstrated a prompt and marked decline of serum levels under therapy with steroids and immunosuppressants in all cases. Treatment regimens included intravenous methylprednisolone (IVMP) in combination with azathioprine, dexamethasone, rituximab, mitoxantrone or prednisolone. AQP4-Ab levels decreased by 4–19% (median 8%) per week; when taking into account only those samples that were taken within the first month after relapse, a decremental rate of 9.3% per week was found. In two patients treated with a combination of azathioprine and prednisolone following IVMP for acute relapse, further samples were obtained during remission, demonstrating constantly low serum levels over more than 350 and 500 days, respectively.
Correlation of CD19 counts and AQP4-Ab levels under treatment with rituximab
CD19 cell numbers declined after treatment with rituximab following a protocol proposed by Cree and co-workers (Cree et al., 2005). They reappeared after 251, 258, 265, 272 and 350 days, respectively, following the last application of rituximab, with slight rises in cell counts being associated with a strong increase in AQP4-Ab values (Fig. 2A, day 3168; Fig. 2B, day 1392). Although application of the drug was followed by a prompt and marked decline in titres (51–90%; P = 0.02, Wilcoxon's matched-pairs rank sum test; Fig. 4), AQP4-Ab remained detectable during therapy with rituximab in 29/30 samples. Moreover, AQP4-Ab was still positive despite the cell numbers being below the detection limit in 16/17 samples. No correlation between CD19 cell count and AQP4-Ab values was found in those patients not treated with rituximab (data not shown).

AQP4-Ab serum levels before and after (values refer to Nadir values) eight applications of rituximab in four patients with NMO/LETM (P = 0.0156; Wilcoxon matched-pairs rank sum test).
Reduced relapse rate under immunsuppressive therapy
Relapse rates in patients treated with rituximab are given in Table 1; median relapse rate was 2.3/year (1.55–2.79) before and 0.51/year (0.46–1.04) after initiation of therapy. Although the overall relapse rate declined under rituximab, at least one relapse occurred in each patient while under therapy (Fig. 2A–C and I). In three of four patients relapses occurred 260, 311 and 364 days, respectively, from last infusion (Fig. 2A–C). Relapses were preceded or paralleled by reoccurrence of CD19 cells and an up to 3-fold rise in AQP4-Ab levels in these cases. Therapy intervals were shortened in two of them resulting in absence of further attacks (Fig. 2A and B). In another patient with elevated AQP4-Ab levels prior to application, rituximab could not prevent clinical attacks 27 and 99 days later (Fig. 2I).
Relapse rates under therapy with various immunosuppressants
| Relapses before initiation of therapy | Relapses after initiation of therapy | ||||
|---|---|---|---|---|---|
| a. Rituximab | |||||
| Pat.1 | 12/2823 days | 1.55/year | 1/795 days | 0.46/yeara | ↓ |
| Pat.2 | 16/2095 days | 2.79/year | 1/889 days | 0.41/yeara | ↓ |
| Pat.3 | 7/1097 days | 2.33/year | 1/647 days | 0.56/yeara | ↓ |
| Pat.4 | 6/964 days | 2.27/year | 2/699 days | 1.04/yeara | ↓ |
| b. IVIg+plasmaexchange | |||||
| Pat.3 | 5/545 days | 3.35/year | 2/395 days | 1.84/year | ↓ |
| c. Mitoxantrone | |||||
| Pat.1 | 6/1611 days | 1.36/year | 3/475 days | 2.31/year | ↑ |
| Pat.4 | 2/309 days | 2.36/year | 1/289 days | 1.26/year | ↓ |
| Pat.5 | 6/979 days | 2.24/year | 3/253 days | 4.33/year | ↑ |
| d. Azathioprine (+prednisolone) | |||||
| Pat.6 | 3/638 days | 1.72/year | 0/710 days | 0/year | ↓ |
| and 0/313 days | 0/yearb | ↓ | |||
| Pat.7 | 4/197 days | 7.41/year | 0/311 days | 0/year | ↓ |
| and 1/499 days | 0.73/yearb | ↓ | |||
| Pat.8 | 3/1293 days | 0.85/year | 1/602 days | 0.61/year | ↔ |
| e. Cyclophosphamide | |||||
| Pat.5 | 10/1295 days | 2.82/year | 1/1610 days | 0.23/year | ↓ |
| f. Interferon beta | |||||
| Pat.1 | 4/1255 days | 1.16/year | 2/343 days | 2.13/year | ↑ |
| Pat.2 | 1/122 days | 2.99/year | 8/937 days | 3.12/year | ↔ |
| Pat.3 | 4/425 days | 3.44/year | 1/102 days | 3.57/year | ↔ |
| g. Copaxone | |||||
| Pat.2 | 10/1091 days | 3.34/year | 2/190 days | 3.84/yearc | ↔ |
| Relapses before initiation of therapy | Relapses after initiation of therapy | ||||
|---|---|---|---|---|---|
| a. Rituximab | |||||
| Pat.1 | 12/2823 days | 1.55/year | 1/795 days | 0.46/yeara | ↓ |
| Pat.2 | 16/2095 days | 2.79/year | 1/889 days | 0.41/yeara | ↓ |
| Pat.3 | 7/1097 days | 2.33/year | 1/647 days | 0.56/yeara | ↓ |
| Pat.4 | 6/964 days | 2.27/year | 2/699 days | 1.04/yeara | ↓ |
| b. IVIg+plasmaexchange | |||||
| Pat.3 | 5/545 days | 3.35/year | 2/395 days | 1.84/year | ↓ |
| c. Mitoxantrone | |||||
| Pat.1 | 6/1611 days | 1.36/year | 3/475 days | 2.31/year | ↑ |
| Pat.4 | 2/309 days | 2.36/year | 1/289 days | 1.26/year | ↓ |
| Pat.5 | 6/979 days | 2.24/year | 3/253 days | 4.33/year | ↑ |
| d. Azathioprine (+prednisolone) | |||||
| Pat.6 | 3/638 days | 1.72/year | 0/710 days | 0/year | ↓ |
| and 0/313 days | 0/yearb | ↓ | |||
| Pat.7 | 4/197 days | 7.41/year | 0/311 days | 0/year | ↓ |
| and 1/499 days | 0.73/yearb | ↓ | |||
| Pat.8 | 3/1293 days | 0.85/year | 1/602 days | 0.61/year | ↔ |
| e. Cyclophosphamide | |||||
| Pat.5 | 10/1295 days | 2.82/year | 1/1610 days | 0.23/year | ↓ |
| f. Interferon beta | |||||
| Pat.1 | 4/1255 days | 1.16/year | 2/343 days | 2.13/year | ↑ |
| Pat.2 | 1/122 days | 2.99/year | 8/937 days | 3.12/year | ↔ |
| Pat.3 | 4/425 days | 3.44/year | 1/102 days | 3.57/year | ↔ |
| g. Copaxone | |||||
| Pat.2 | 10/1091 days | 3.34/year | 2/190 days | 3.84/yearc | ↔ |
↓/↑ = change > 0.5 relapses/year, ↔ = change <= 0.5 relapses/year.
aPatients were treated with various immunomodulatory and/or immunosuppressive agents prior to rituximab. bAfter interruption of therapy, which had resulted in one new relapse. cAllowing for six months latency of action (otherwise 2/370 days or 1.98/year).
Relapse rates under therapy with various immunosuppressants
| Relapses before initiation of therapy | Relapses after initiation of therapy | ||||
|---|---|---|---|---|---|
| a. Rituximab | |||||
| Pat.1 | 12/2823 days | 1.55/year | 1/795 days | 0.46/yeara | ↓ |
| Pat.2 | 16/2095 days | 2.79/year | 1/889 days | 0.41/yeara | ↓ |
| Pat.3 | 7/1097 days | 2.33/year | 1/647 days | 0.56/yeara | ↓ |
| Pat.4 | 6/964 days | 2.27/year | 2/699 days | 1.04/yeara | ↓ |
| b. IVIg+plasmaexchange | |||||
| Pat.3 | 5/545 days | 3.35/year | 2/395 days | 1.84/year | ↓ |
| c. Mitoxantrone | |||||
| Pat.1 | 6/1611 days | 1.36/year | 3/475 days | 2.31/year | ↑ |
| Pat.4 | 2/309 days | 2.36/year | 1/289 days | 1.26/year | ↓ |
| Pat.5 | 6/979 days | 2.24/year | 3/253 days | 4.33/year | ↑ |
| d. Azathioprine (+prednisolone) | |||||
| Pat.6 | 3/638 days | 1.72/year | 0/710 days | 0/year | ↓ |
| and 0/313 days | 0/yearb | ↓ | |||
| Pat.7 | 4/197 days | 7.41/year | 0/311 days | 0/year | ↓ |
| and 1/499 days | 0.73/yearb | ↓ | |||
| Pat.8 | 3/1293 days | 0.85/year | 1/602 days | 0.61/year | ↔ |
| e. Cyclophosphamide | |||||
| Pat.5 | 10/1295 days | 2.82/year | 1/1610 days | 0.23/year | ↓ |
| f. Interferon beta | |||||
| Pat.1 | 4/1255 days | 1.16/year | 2/343 days | 2.13/year | ↑ |
| Pat.2 | 1/122 days | 2.99/year | 8/937 days | 3.12/year | ↔ |
| Pat.3 | 4/425 days | 3.44/year | 1/102 days | 3.57/year | ↔ |
| g. Copaxone | |||||
| Pat.2 | 10/1091 days | 3.34/year | 2/190 days | 3.84/yearc | ↔ |
| Relapses before initiation of therapy | Relapses after initiation of therapy | ||||
|---|---|---|---|---|---|
| a. Rituximab | |||||
| Pat.1 | 12/2823 days | 1.55/year | 1/795 days | 0.46/yeara | ↓ |
| Pat.2 | 16/2095 days | 2.79/year | 1/889 days | 0.41/yeara | ↓ |
| Pat.3 | 7/1097 days | 2.33/year | 1/647 days | 0.56/yeara | ↓ |
| Pat.4 | 6/964 days | 2.27/year | 2/699 days | 1.04/yeara | ↓ |
| b. IVIg+plasmaexchange | |||||
| Pat.3 | 5/545 days | 3.35/year | 2/395 days | 1.84/year | ↓ |
| c. Mitoxantrone | |||||
| Pat.1 | 6/1611 days | 1.36/year | 3/475 days | 2.31/year | ↑ |
| Pat.4 | 2/309 days | 2.36/year | 1/289 days | 1.26/year | ↓ |
| Pat.5 | 6/979 days | 2.24/year | 3/253 days | 4.33/year | ↑ |
| d. Azathioprine (+prednisolone) | |||||
| Pat.6 | 3/638 days | 1.72/year | 0/710 days | 0/year | ↓ |
| and 0/313 days | 0/yearb | ↓ | |||
| Pat.7 | 4/197 days | 7.41/year | 0/311 days | 0/year | ↓ |
| and 1/499 days | 0.73/yearb | ↓ | |||
| Pat.8 | 3/1293 days | 0.85/year | 1/602 days | 0.61/year | ↔ |
| e. Cyclophosphamide | |||||
| Pat.5 | 10/1295 days | 2.82/year | 1/1610 days | 0.23/year | ↓ |
| f. Interferon beta | |||||
| Pat.1 | 4/1255 days | 1.16/year | 2/343 days | 2.13/year | ↑ |
| Pat.2 | 1/122 days | 2.99/year | 8/937 days | 3.12/year | ↔ |
| Pat.3 | 4/425 days | 3.44/year | 1/102 days | 3.57/year | ↔ |
| g. Copaxone | |||||
| Pat.2 | 10/1091 days | 3.34/year | 2/190 days | 3.84/yearc | ↔ |
↓/↑ = change > 0.5 relapses/year, ↔ = change <= 0.5 relapses/year.
aPatients were treated with various immunomodulatory and/or immunosuppressive agents prior to rituximab. bAfter interruption of therapy, which had resulted in one new relapse. cAllowing for six months latency of action (otherwise 2/370 days or 1.98/year).
In the only patient undergoing long-term treatment with cyclophosphamide, 10 relapses occurred within 1295 days (=2.82/year) prior to initiation of therapy but only one within 1610 days (=0.23/year) while on therapy (50 mg/day orally; later reduced to 50 mg eod; Fig. 2G). Long-term treatment with mitoxantrone was tried in three patients but resulted in a reduction of clinical attacks in only one of them (Table 1). However, relapse free periods prior to any long-term immunosuppressive therapy were found to last up to 40 months (median 308.5 days; range 72–1236). Immunmodulatory therapy with interferon beta was not paralleled by decrease in relapse rate (Table 1).
Suspension of azathioprine is followed by increase in AQP4-Ab levels and clinical attack
In three patients, therapy with azathioprine was maintained for more than the maximum latency period of six months (268, 235 and 673 days) and interrupted later in the disease course. Azathioprine resulted in a marked decline in relapse rate in two of them, and a mild decline in one (Table 1). While 10 relapses occurred within 2199 days prior to therapy, only two relapses occurred within 2212 days under therapy. Annualized relapse rates prior to and under treatment are given in the Table 1.
Interruption of therapy was, however, followed by clinical relapse (34, 65 and 181 days after suspension, respectively) and increase of AQP4-Ab values (2.2-, 1.4- and 7.2-fold, respectively) in all cases (Fig. 2D, F, H and J). Relapses occurred despite sustained therapy with low dose prednisolone (tapered off to 5 mg/day one month before relapse) in one patient (Fig. 2D). In the other patients, prednisolone was interrupted 155 and 302 days, respectively, prior to interruption of azathioprine without causing clinical deterioration or relapse (Fig. 2F, H and J).
High AQP4-Ab levels are not always associated with clinical relapse
Despite the observations above, a confirmed and considerable rise in AQP4-Ab levels (284%, 149% and 95%, respectively, compared with last Nadir value), in three patients was not followed by clinical relapse (Fig. 2C, F and G). In one of them, rituximab-induced B-cell depletion interrupted any further increase of antibody levels (Fig. 2C). In the two other patients, AQP4-Ab levels rose continuously over a period of 956 and 263 days, respectively, when reducing the dosage of cyclophosphamide or azathioprine without resulting in clinical deterioration or relapse (Fig. 2F and G). Despite the increasing AQP4-Ab levels, the relapse rate declined under therapy with cyclophosphamide to one relapse within 1610 days compared with 10 relapses within 1295 days before therapy was initiated.
Discussion
In this study, we provide quantitative data on AQP4-Ab in the long-term course of NMO. Our finding that relapses in NMO is preceded by a rise in serum AQP4-Ab levels strengthens the case for the antibody being involved in the pathogenesis of the disease. Although the correlation found in our study does not prove a causal relationship on its own, it is consistent with recent evidence from histopathological and immunological studies that indicate a direct contribution of AQP4-Ab to tissue damage in NMO (Hinson et al., 2007; Misu et al., 2007; Roemer et al., 2007; Jarius et al., 2008; Waters et al., 2008). This is in contrast to other antibody-associated autoimmune diseases of the CNS (such as paraneoplastic neurological disorders), in which the antibodies are considered mere diagnostic markers with no well accepted role in the mechanisms of lesion pathology.
AQP4-Ab was detectable in serum during relapse as well as during remission, both in untreated patients and in almost all samples obtained under immunosuppressive therapy, suggesting that AQP4-Ab testing can be of diagnostic relevance independently of disease activity or treatment status.
Shortly before relapse, AQP4-Ab levels rose rapidly (∼20% per week) and markedly (up to ∼290%) in all cases studied. In addition, in one of our patients, we found that AQP4-Ab levels rose selectively during clinical attack despite immunosuppressive treatment, while three other auto-antibodies, unrelated to NMO, declined at the same time or remained low. These findings argue in favour of the increase in AQP4-Ab levels being specific in this patient and against it being simply part of a general increase in B-cell activity during relapse.
Interestingly, however, no general threshold value for triggering clinical relapse was found, but serum levels detected during relapse differed widely both intra- and inter-individually. Although all attacks studied were paralleled by a rise in AQP4-Ab levels, in a minority of samples titres during remission were found to be higher at some time-point than titres found during relapse in the same patient or other patients. While in some cases, low titres were associated with clinical relapse, high titres were not paralleled by clinical disease activity in some patients treated with immunosuppressants. These observations are similar to those in myasthenia gravis, an accepted antibody-mediated neurological disease, and do not argue against the hypothesis that the antibodies are pathogenic. However, they indicate that the presence of AQP4-Ab alone may not be sufficient to cause disease; other factors, for instance, disease-specific T cells, raised cytokines, unspecific stimulation by exogenous triggers or damage to the blood brain barrier, might be required to initiate or cause tissue damage. Our results do suggest, however, that AQP4-Ab will generally be of use as a marker of disease activity over time in individual patients.
Initiation of immunosuppression was apparently followed by a marked and rapid decline of both AQP4-Ab levels and flare rates in all of our patients. However, taking into account the well known latency of action of azathioprine and cyclophosphamide this prompt response was probably rather caused by IVMP, which was applied as treatment for acute relapse. Importantly, however, antibody levels remained low under combined therapy with azathioprine and prednisolone over an observation period of up to 500 days with no further clinical attacks, while interruption of azathioprine resulted in clinical relapse and an increase of antibody levels within 1–6 months. These findings are in accordance with results from a small Japanese study, reporting a marked and sustained decline of AQP4-Ab titres in two patients undergoing treatment with azathioprine and prednisolone with no relapse over 6 and 11 months, respectively (Takahashi et al., 2007).
Cyclophosphamide (50 mg/day) resulted in a long-lasting relapse-free interval in one of our patients, with 10 relapses within 1295 days (2.82/year) prior to initiation of therapy but only one within 1610 days (0.23/year) under therapy. However, a continuous yet slightly oscillating increase in AQP4-Ab titres was found after dose reduction, which was not followed by clinical relapse after 956 days. This again indicates that the antibody might not be sufficient to cause clinical relevant damage on its own, but further players might be involved in the pathogenesis of NMO.
While azathioprine (and prednisolone) was most effective in lowering the relapse rate in our patients (Table 1), treatment with rituximab, a therapeutic monoclonal antibody causing temporary B-cell depletion (Kazkaz and Isenberg, 2004), resulted in the most pronounced decline in AQP4-Ab levels. Nonetheless, AQP4-Ab did not fall below cut-off in almost all samples obtained under rituximab. Moreover, AQP4-Ab remained positive despite the CD19 cell counts being below the detection limit. There are two possible explanations for this finding: Although rituximab causes complete depletion of CD19-positive peripheral B cells, it does not affect plasma cells. Second, although rituximab interrupts the steady flow of new plasma cells from differentiating B cells, the so-called long-life plasma cells, some of which may survive for the lifespan of the host, could maintain specific antibody production over long periods. Our findings are in line with previous studies demonstrating a decline of specific autoantibodies under therapy with rituximab in such well recognized autoimmune disorders such as Graves disease, immune thrombocytopenic purpura or pemphigus vulgaris (Levesque and St Clair, 2008).
It is important to notice that the duration of action varied considerably among patients treated with rituximab. CD19 cells reappeared in our patients 250–350 days after last infusion. Fixed therapy intervals might thus not be advisable, but application intervals might have to be adapted individually based on CD19 counts.
Importantly, the reappearance of even low B-cell numbers was found to be sufficient to induce an increase in AQP4-Ab values, and reoccurrence of CD19 cells was associated with a high relapse risk. CD19 counting might thus be an alternative to AQP4-Ab testing in those patients. However, B cells remained detectable in all patients not treated with rituximab, with no correlation between CD19 counts and AQP4-Ab serum values.
There are some obvious limitations of this study, including the small number of patients analysed, the possibility of unintentional selection bias and the wide variety of treatments regimens used. These result from its retrospective design, the rarity of the disease investigated, and the short availability of AQP4-Ab testing. However, we believe that the large number of samples investigated, the long observation periods in each patient, and the detailed clinical data adds to the growing body of evidence for a significant relationship between AQP4-Ab levels and clinical state.
In conclusion, this study shows that AQP4 antibodies are higher overall in patients during relapse than during remission, demonstrates a correlation between rises in antibody levels and clinical attacks, and illustrates the decline of AQP4-Ab levels during various immunosuppressive therapies which were associated with reduced relapse rates. Although only four patients were treated with rituximab, there were marked falls in AQP4 antibodies correlating with the changes in CD19 positive cells, however the duration of action of rituximab was quite variable between patients. Overall, these findings strengthen the case for the role of AQP4-Ab in the pathogenesis of NMO/LETM. Larger studies are now warranted to affirm and to extend our findings in a prospective and controlled setting.
Funding
European Neurological Society (ENS) (to S.J.); European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) (to S.J.); University of Cologne (Prof. R. Voltz), Germany (to S.J.); NIHR Biomedical Research Centre Programme (to P.W. and A.V.). The funding sources had no role in study design; collection, analysis or interpretation of data; writing of the paper or decision to submit it for publication.
References
- Ab
antibody
- AChR
acetylcholine receptor
- AQP4
aquaporin-4
- CNS
central nervous system
- FIPA
fluorescence based immunoprecipitation assay
- FU
fluorescence units
- IgG
immunoglobulin G
- IVMP
intravenous methylprednisolone
- LETM
longitudinally extensive transverse myelitis
- MRI
magnetic resonance imaging
- NMO
neuromyelitis optica
- TG
thyroglobulin
- TPO
thyroid peroxidase


{kind=link}
{kind=link}
{kind=link}
{kind=link}