




Abstract
Among patients with frontotemporal lobar degeneration (FTLD), the respective frequencies of dominant 17q21-linked tau-negative FTLD (with unidentified molecular defect) and 17q21-linked tau-positive FTLD (due to MAPT mutations) remain unknown. Here, in a series of 98 genealogically unrelated Belgian FTLD patients, we identified an ancestral 8 cM MAPT containing haplotype in two patients belonging to multiplex families DR2 and DR8, without demonstrable MAPT mutations, in which FTLD was conclusively linked to 17q21 [maximum summed log of the odds (LOD) score of 5.28 at D17S931]. Interestingly, the same DR2–DR8 ancestral haplotype was observed in five additional familial FTLD patients, indicative of a founder effect. In the FTLD series, the DR2–DR8 ancestral haplotype explained 7% (7 out of 98) of FTLD and 17% (7 out of 42) of familial FTLD and was seven times more frequent than MAPT mutations (1 out of 98 or 1%). Clinically, DR2–DR8 haplotype carriers presented with FTLD often characterized by language impairment, and in one carrier the neuropathological diagnosis was FTLD with rare tau-negative ubiquitin-positive inclusions. Together, these results strongly suggest that the DR2–DR8 founder haplotype at 17q21 harbours a tau-negative FTLD causing mutation that is a much more frequent cause of FTLD in Belgium than MAPT mutations.
Introduction
In the age group below 65 years, frontotemporal lobar degeneration (FTLD) [MIM 600274] comprises 12–20% of demented patients and is the second-most common form of neurodegenerative presenile dementia after Alzheimer's disease (AD) [MIM 104300] (Neary et al., 1998; Ratnavalli et al., 2002; Harvey et al., 2003). Clinically, FTLD is characterized by progressive degeneration of frontal and/or temporal brain regions leading to behavioural and personality disturbances including disinhibition, perseveration and emotional blunting, often accompanied by progressive language dysfunctions, and eventually evolves into general cognitive impairment (Neary et al., 1998).
A positive family history of dementia is present in 38–50% of FTLD patients, and in the majority of the families the disease is inherited in an autosomal dominant manner (Stevens et al., 1998; Chow et al., 1999; Poorkaj et al., 2001a; Rosso et al., 2003). Linkage studies have identified FTLD loci on chromosomes 3 [MIM 600795] (Brown et al., 1995), 9 [MIM 105550] (Hosler et al., 2000) and 17 (Foster et al., 1997). Besides a very recently identified mutation in the charged multi-vesicular body protein 2B (CHMP2B) at 3p11 (Skibinski et al., 2005), all other known FTLD mutations affect the microtubule-associated protein tau (MAPT [MIM 157140]) at 17q21 (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). To date, 38 different MAPT mutations have been identified in 111 dementia families worldwide (Rademakers et al., 2004; AD&FTD Mutation database: http://www.molgen.ua.ac.be/ADMutations). It was estimated that MAPT mutations explain 5–20% of FTLD in general and 10–43% of familial FTLD (Rizzu et al., 1999; Poorkaj et al., 2001a; Rosso et al., 2003). Neuropathologically, MAPT mutation carriers are characterized by intraneuronal and/or glial tau-positive inclusions (tauopathy). More recent genetic and clinicopathological studies, however, demonstrated that the majority of FTLD patients could not be explained by MAPT mutations and lacked tau pathology (tau-negative FTLD) (Rizzu et al., 1999; Mann et al., 2000; Morris et al., 2001; Poorkaj et al., 2001a; Rosso et al., 2003; Hodges et al., 2004; Johnson et al., 2005). Surprisingly, several tau-negative FTLD families have been conclusively linked to a region at 17q21 that contains MAPT (Rademakers et al., 2004). In three conclusively 17q21-linked families, the neuropathological phenotype has been described as either ‘dementia lacking distinctive histopathology’ (DLDH) (Lendon et al., 1998) or ‘FTLD with tau-negative and ubiquitin-positive inclusions’ (FTDU) (Rosso et al., 2001; Rademakers et al., 2002). In the highly informative Dutch family, 1083, we reduced the candidate region for FTLD at 17q21 to a 4.8 cM interval encompassing MAPT (Rademakers et al., 2002). Recently, we excluded mutations in MAPT by genomic sequencing of 138.5 kb in 17q21-linked tau-negative FTLD patients from family 1083 and Dutch III (Cruts et al., 2005). In addition, we and others showed that MAPT is surrounded by three highly homologous low-copy repeats (LCRs) in a region of 1.7 Mb. These LCRs induced a genomic inversion polymorphism explaining the high degree of linkage disequilibrium in the MAPT genomic region resulting in two extended haplotypes H1 and H2 (Baker et al., 1999; Cruts et al., 2005; Stefansson et al., 2005). The presence of multiple homologous LCRs in the region could be responsible for more complex genomic rearrangements that underlie 17q21-linked tau-negative FTLD (Stankiewicz and Lupski, 2002). However, so far the molecular defect of tau-negative FTLD remains unknown (Cruts et al., 2005), and an estimation of its frequency is currently difficult.
Here, we performed a detailed molecular genetic, clinical and pathological study of 17q21-linked tau-negative FTLD in Belgium. We present convincing evidence that supports a tau-negative FTLD founder haplotype harbouring a causal mutation, which is responsible for a substantial fraction of familial FTLD. Our data also indicated that this mutation is a much more important cause of FTLD than MAPT mutations.
Materials and methods
Patients, families and controls
A total of 98 FTLD patients was derived from a prospective Belgian study of neurodegenerative and vascular dementia (n = 62) (Engelborghs et al., 2003) and from a collection of demented patients referred to our Molecular Diagnostic Unit for molecular genetic testing (n = 36) (Table 1). The local medical ethical committee approved the prospective Belgian study of neurodegenerative and vascular dementia and all participating individuals gave written informed consent. Diagnosis of FTLD was reached in consensus by at least two neurologists using established clinical criteria (Neary et al., 1998). All patients underwent neuroimaging (CT-scan and/or MRI) and neuropsychological testing. FTLD patients from the Molecular Diagnostic Unit series were selected on the basis of a clinical diagnosis of FTLD and medical records provided by the referring neurologist or gerontologist. None of the 98 FTLD patients was known to be genealogically related. A neuropathological diagnosis was available for seven patients. The ascertainment of Belgian control individuals (n = 181) was described previously (Pals et al., 2004).
General descriptives of Belgian FTLD patients
| Male : female ratio | Mean age at onset (range) | Positive family history (%)a | |
|---|---|---|---|
| Prospective dementia study (n = 62) | 1.21 | 65.1 (40–90) | 35 |
| Molecular Diagnostic Unit (n = 36) | 1.25 | 56.5 (18–74) | 56 |
| Total FTLD patients (n = 98) | 1.23 | 61.4 (18–90) | 43 |
| Male : female ratio | Mean age at onset (range) | Positive family history (%)a | |
|---|---|---|---|
| Prospective dementia study (n = 62) | 1.21 | 65.1 (40–90) | 35 |
| Molecular Diagnostic Unit (n = 36) | 1.25 | 56.5 (18–74) | 56 |
| Total FTLD patients (n = 98) | 1.23 | 61.4 (18–90) | 43 |
Positive family history was defined as having at least one first degree relative with dementia.
General descriptives of Belgian FTLD patients
| Male : female ratio | Mean age at onset (range) | Positive family history (%)a | |
|---|---|---|---|
| Prospective dementia study (n = 62) | 1.21 | 65.1 (40–90) | 35 |
| Molecular Diagnostic Unit (n = 36) | 1.25 | 56.5 (18–74) | 56 |
| Total FTLD patients (n = 98) | 1.23 | 61.4 (18–90) | 43 |
| Male : female ratio | Mean age at onset (range) | Positive family history (%)a | |
|---|---|---|---|
| Prospective dementia study (n = 62) | 1.21 | 65.1 (40–90) | 35 |
| Molecular Diagnostic Unit (n = 36) | 1.25 | 56.5 (18–74) | 56 |
| Total FTLD patients (n = 98) | 1.23 | 61.4 (18–90) | 43 |
Positive family history was defined as having at least one first degree relative with dementia.
For molecular genetic studies, FTLD index patients or a living relative was contacted by research nurses under the supervision of a physician experienced in clinical and molecular genetics of dementia. Detailed information on family history of dementia was gathered and additional patients and unaffected family members were asked to participate in genetic studies. With written informed consent blood samples were collected for DNA extraction and the establishment of lymphoblast cell lines. Informed consent was also requested for autopsy and neuropathological examination. The local medical ethical committee of the University of Antwerp approved the research protocols for molecular genetic and neuropathological studies. For phase-determined haplotype frequency estimation in healthy controls, we used 92 DNA samples from 23 healthy tetrads of Belgian origin, each consisting of parents and two children.
Mutation analysis
MAPT mutation analysis was performed on genomic DNA of the 98 FTLD patients by direct sequencing of exons 1 and 9 to 13. In addition, FTLD patients derived from the Belgian prospective dementia study and 12 FTLD patients from the Molecular Diagnostic Unit, including the index patients of family DR2 (III-6) and DR8 (III-28), were also screened for mutations in the coding exons 3–12 of the presenilin-1 gene (PSEN1 [MIM 104311]) and presenilin-2 gene (PSEN2 [MIM 600759]), the amyloid precursor protein gene (APP [MIM 104760]) coding exons 16 and 17, and coding exon 2 of the prion protein gene (PRNP [MIM 176640]). More extensive mutation analysis of MAPT was performed in three individuals of DR2 (two patients: III-8, III-6; one control: III-22) and DR8 (two patients: III-28, III-32; one control: III-24) and included all MAPT exons and intron 13 that is retained in human MAPT transcripts (Poorkaj et al., 2001b). Standard 20 μl polymerase chain reaction (PCR) amplifications on genomic DNA with empirically defined optimal annealing temperatures were performed and amplification products were purified. Purified products were sequenced in both directions using the BigDye Terminator Cycle Sequencing kit v3.1 (Applied Biosystems, Foster City, CA, USA) and analysed on an ABI3730 automated sequencer (Applied Biosystems, Foster City, CA, USA).
STR genotyping and MAPT H1–H2 haplotyping
For linkage studies, 77 DR2 and DR8 family members (Fig. 1) were genotyped using 18 chromosome 17q21 fluorescently labelled STR markers spanning a 17.7 cM region between D17S1863 and D17S1795. Fifteen STR markers were selected from the Marshfield gender-averaged genetic map, and three novel STR markers, Chr17-16, Chr17-19 and Chr17-43, were identified with the Tandem Repeats Finder program (Benson, 1999) (Table 2). Chr17-16 is located in AC008105.32 starting at nt 82045, Chr17-19 is located in AC091628.2 at nt 35879 and Chr17-43 is located in in AC068234.8 at nt 138430. For allele sharing analysis in the 98 FTLD patients, the relatives of DR2–DR8 ancestral haplotype carriers and the 23 healthy Belgian tetrads, 14 of the 18 STR markers were selected for genotyping. Allele frequencies were calculated in 92 control chromosomes derived from the parents of the Belgian tetrads. Genomic DNA (20 ng) was amplified in 20 μl multiplex PCRs at annealing temperature of 58°C by use of fluorescently labelled primers. PCR products were sized on an ABI 3730 automated sequencer (Applied Biosystems), and genotypes were assigned using in-house developed genotyping software.
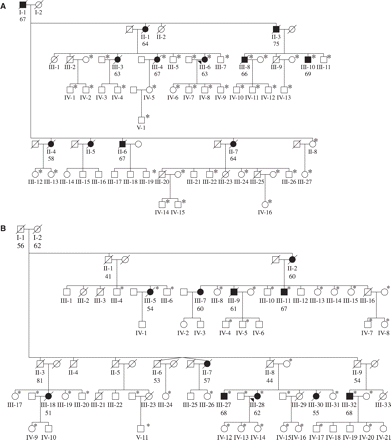
Pedigrees of Belgian FTLD families DR2 (A) and DR8 (B). Filled symbols represent FTLD patients; open symbols represent unaffected individuals. The number below the individuals denotes age at onset for patients and age at death for obligate carriers. When available, affection status and onset age were determined from medical records (III-4, III-6, III-8 and III-10 of family DR2, and III-9, III-18 and III-28 of family DR8), for the other patients these were determined using information provided by family informants. An arrowhead indicates the index patient. An asterisk (*) denotes that DNA was available for genotyping.
Two-point LOD scores at 17q21 in Belgian FTLD families DR2 and DR8
| Marker | Physical distance (Mb) | Genetic distance (cM) | LOD score at 𝛉 = 0 | ||
|---|---|---|---|---|---|
| DR2 | DR8 | ||||
| D17S1863 | 0 | 0 | −1.47 | −0.02 | |
| D17S1818 | 8.47 | 9.7 | 0.2 | 2.26 | |
| D17S1814 | 9.42 | 10.7 | 0.81 | 0.00 | |
| D17S800 | 10.36 | 11.3 | 0.17 | 2.79 | |
| D17S1787 | 11.03 | 11.3 | 0.78 | 0.4 | |
| D17S1793 | 11.66 | 12.4 | −0.06 | 0.67 | |
| D17S902 | 13.05 | 13.4 | 1.42 | 2.37 | |
| D17S951 | 13.23 | 12.9 | 0.3 | 2.79 | |
| D17S1861 | 14.21 | 12.9 | 1.01 | 2.47 | |
| D17S934 | 14.46 | 12.9 | 0.71 | 2.88 | |
| Chr17-16 | 14.73 | – | −0.12 | 1.81 | |
| D17S810 | 14.89 | 12.9 | 0.38 | 0.59 | |
| Chr17-19 | 15.41 | – | 0.58 | 0.95 | |
| D17S920 | 16.22 | 13.4 | 0.7 | 0.41 | |
| D17S931 | 16.4 | 16.1 | 1.45 | 3.32 | |
| Chr17-43 | 16.73 | – | 1.47 | 1.76 | |
| D17S1785 | 17.43 | 16.1 | 0.47 | 2.6 | |
| D17S1795 | 19.33 | 17.7 | −1.06 | 2.72 | |
| Marker | Physical distance (Mb) | Genetic distance (cM) | LOD score at 𝛉 = 0 | ||
|---|---|---|---|---|---|
| DR2 | DR8 | ||||
| D17S1863 | 0 | 0 | −1.47 | −0.02 | |
| D17S1818 | 8.47 | 9.7 | 0.2 | 2.26 | |
| D17S1814 | 9.42 | 10.7 | 0.81 | 0.00 | |
| D17S800 | 10.36 | 11.3 | 0.17 | 2.79 | |
| D17S1787 | 11.03 | 11.3 | 0.78 | 0.4 | |
| D17S1793 | 11.66 | 12.4 | −0.06 | 0.67 | |
| D17S902 | 13.05 | 13.4 | 1.42 | 2.37 | |
| D17S951 | 13.23 | 12.9 | 0.3 | 2.79 | |
| D17S1861 | 14.21 | 12.9 | 1.01 | 2.47 | |
| D17S934 | 14.46 | 12.9 | 0.71 | 2.88 | |
| Chr17-16 | 14.73 | – | −0.12 | 1.81 | |
| D17S810 | 14.89 | 12.9 | 0.38 | 0.59 | |
| Chr17-19 | 15.41 | – | 0.58 | 0.95 | |
| D17S920 | 16.22 | 13.4 | 0.7 | 0.41 | |
| D17S931 | 16.4 | 16.1 | 1.45 | 3.32 | |
| Chr17-43 | 16.73 | – | 1.47 | 1.76 | |
| D17S1785 | 17.43 | 16.1 | 0.47 | 2.6 | |
| D17S1795 | 19.33 | 17.7 | −1.06 | 2.72 | |
Maximal two-point LOD scores are indicated in bold. For all STR markers observed alleles were assigned equal allele frequencies. Physical distances of the STR markers were deduced from the UCSC Human Genome Browser. Genetic positions of the STR markers were obtained from the Marshfield gender-averaged map.
Two-point LOD scores at 17q21 in Belgian FTLD families DR2 and DR8
| Marker | Physical distance (Mb) | Genetic distance (cM) | LOD score at 𝛉 = 0 | ||
|---|---|---|---|---|---|
| DR2 | DR8 | ||||
| D17S1863 | 0 | 0 | −1.47 | −0.02 | |
| D17S1818 | 8.47 | 9.7 | 0.2 | 2.26 | |
| D17S1814 | 9.42 | 10.7 | 0.81 | 0.00 | |
| D17S800 | 10.36 | 11.3 | 0.17 | 2.79 | |
| D17S1787 | 11.03 | 11.3 | 0.78 | 0.4 | |
| D17S1793 | 11.66 | 12.4 | −0.06 | 0.67 | |
| D17S902 | 13.05 | 13.4 | 1.42 | 2.37 | |
| D17S951 | 13.23 | 12.9 | 0.3 | 2.79 | |
| D17S1861 | 14.21 | 12.9 | 1.01 | 2.47 | |
| D17S934 | 14.46 | 12.9 | 0.71 | 2.88 | |
| Chr17-16 | 14.73 | – | −0.12 | 1.81 | |
| D17S810 | 14.89 | 12.9 | 0.38 | 0.59 | |
| Chr17-19 | 15.41 | – | 0.58 | 0.95 | |
| D17S920 | 16.22 | 13.4 | 0.7 | 0.41 | |
| D17S931 | 16.4 | 16.1 | 1.45 | 3.32 | |
| Chr17-43 | 16.73 | – | 1.47 | 1.76 | |
| D17S1785 | 17.43 | 16.1 | 0.47 | 2.6 | |
| D17S1795 | 19.33 | 17.7 | −1.06 | 2.72 | |
| Marker | Physical distance (Mb) | Genetic distance (cM) | LOD score at 𝛉 = 0 | ||
|---|---|---|---|---|---|
| DR2 | DR8 | ||||
| D17S1863 | 0 | 0 | −1.47 | −0.02 | |
| D17S1818 | 8.47 | 9.7 | 0.2 | 2.26 | |
| D17S1814 | 9.42 | 10.7 | 0.81 | 0.00 | |
| D17S800 | 10.36 | 11.3 | 0.17 | 2.79 | |
| D17S1787 | 11.03 | 11.3 | 0.78 | 0.4 | |
| D17S1793 | 11.66 | 12.4 | −0.06 | 0.67 | |
| D17S902 | 13.05 | 13.4 | 1.42 | 2.37 | |
| D17S951 | 13.23 | 12.9 | 0.3 | 2.79 | |
| D17S1861 | 14.21 | 12.9 | 1.01 | 2.47 | |
| D17S934 | 14.46 | 12.9 | 0.71 | 2.88 | |
| Chr17-16 | 14.73 | – | −0.12 | 1.81 | |
| D17S810 | 14.89 | 12.9 | 0.38 | 0.59 | |
| Chr17-19 | 15.41 | – | 0.58 | 0.95 | |
| D17S920 | 16.22 | 13.4 | 0.7 | 0.41 | |
| D17S931 | 16.4 | 16.1 | 1.45 | 3.32 | |
| Chr17-43 | 16.73 | – | 1.47 | 1.76 | |
| D17S1785 | 17.43 | 16.1 | 0.47 | 2.6 | |
| D17S1795 | 19.33 | 17.7 | −1.06 | 2.72 | |
Maximal two-point LOD scores are indicated in bold. For all STR markers observed alleles were assigned equal allele frequencies. Physical distances of the STR markers were deduced from the UCSC Human Genome Browser. Genetic positions of the STR markers were obtained from the Marshfield gender-averaged map.
For determination of the MAPT extended haplotypes H1 and H2 (Baker et al., 1999), the MAPT SNP16 (g.8117G > A; numbering according to GenBank accession number AC091628.2) was genotyped in the 98 FTLD patients and family members of the FTLD families DR2 and DR8 (Rademakers et al., 2005).
Linkage analysis
Two-point and multi-point log of the odds (LOD) scores were calculated using MLINK and LINKMAP from the LINKAGE software package version 5.2 (Lathrop et al., 1985). We assumed an autosomal dominant inheritance model with reduced age-dependent penetrance for the trait locus. The estimated population frequency of the disease gene was set at 0.001. Nine liability classes for disease penetrance, based on the cumulative risk curve calculated from the mean onset age for dementia in each family, with a maximal disease penetrance of 90% when older than 85 years, were used. Phenocopy rates were also age-dependent (Ott et al., 1995).
Neuropathological and immunohistochemical analysis
A post-mortem neuropathological study was performed on patient DR31.1. The brain was fixed for 3 months in 10% formalin, and classic staining techniques for myelin, cytology, fibrillary glia, neutral fats and lipopigments (periodic acid-Schiff; PAS) were performed on coronal 30 μm hemispheric sections sliced on a freezing microtome from two frontotemporal lobe regions: at the level of the amygdala and more posterior at the level of the rostral thalamus. Paraffin-embedded blocks were also prepared from the superior frontal gyrus, cingular gyrus, superior temporal gyrus, hippocampus, parahippocampal gyrus, occipital gyrus, midbrain at the level of the substantia nigra, pons and cerebellum. Sections 5 μm thick were examined by routine histopathological methods and immunostained with the following antibodies: AT8 directed against hyperphosphorylated protein tau (Innogenetics, Zwijnaarde, Belgium), 4G8 against residues 18–24 of Aβ (Signet, Dedham, MA, USA) and ubiquitin (Dako, Glostrup, Denmark). Immunohistochemistries for these antibodies were performed as described previously (Kumar-Singh et al., 2002).
Results
Belgian FTLD patient series
Molecular genetic analysis of five dementia genes—APP, PSEN1, PSEN2, MAPT and PRNP—in 98 Belgian FTLD patients (Table 1) showed a mutation in only two patients. One is the familial patient, which we reported previously (Dermaut et al., 2004), who carried a PSEN1 Gly183Val mutation and had pathologically confirmed Pick's disease. In the second patient we identified in this study a novel MAPT mutation in exon 9 (g110065 G > A, gDNA numbering is relative to AC091628.2 starting at nt.1) predicting an amino acid substitution at codon 273 (MAPT Gly273Arg). MAPT Gly273Arg affects the most C-terminal and highly conserved residue of the first microtubule-binding domain of tau. Age at onset in this patient was 63 years and the disease started with memory disturbances evolving in FTLD with parkinsonism. The MAPT Gly273Arg mutation was absent in 181 Belgian control individuals. No mutations in APP, PSEN2 or PRNP were observed.
In the entire FTLD patient series, we obtained autopsy data for seven patients: four patients had a diagnosis of FTDU, one patient had DLDH, one had AD and one had Pick's disease, the latter one being the PSEN1 Gly183Val mutation carrier.
Belgian FTLD families DR2 and DR8
Two FTLD families DR2 and DR8 were ascertained through two index patients from the Molecular Diagnostic Unit FTLD series that had no mutation in MAPT and a positive family history suggestive of autosomal dominant transmission of dementia. For genetic linkage studies we collected blood samples from 38 and 39 patients and relatives from DR2 and DR8, respectively (Fig. 1A and B). Affection status and onset age were determined using information provided by family informants and medical records when available. The mean age at onset in family DR2 was 65.73 years (range 58–75 years) and 60.27 years (range 51–68 years) in family DR8.
We selected 18 STR markers covering the minimal FTLD candidate region at 17q21 (Rademakers et al., 2002), to test linkage in the two multiplex FTLD families DR2 and DR8. For family DR2, two-point LOD scores > 1 were calculated for 4 of the 18 STR markers, with the highest LOD score of 1.47 at marker Chr17-43 (recombination fraction, 𝛉 = 0) (Table 2). For family DR8, nine LOD scores > 2 were calculated with one conclusive LOD score of 3.32 at marker D17S931 (𝛉 = 0) (Table 2). Multi-point linkage analysis raised the maximum LOD scores to 3.49 in family DR8 and 1.79 in family DR2 in the interval D17S920-D17S931-D17S1795 on top of D17S931 (𝛉 = 0) (data not shown).
In order to confirm linkage to chromosome 17q21 in families DR2 and DR8 and to delineate a minimal candidate region on the basis of meiotic recombination events, haplotypes were reconstructed from genotype data of the 18 markers (Fig. 2A and B). In family DR8 all patients carried the complete risk haplotype including individuals (II-1, II-3, II-8 and II-9) who were obligate carriers. Both founders (I-1 and I-2) had died before or within the onset age range of dementia in the family. In family DR2, a risk haplotype was also observed in all patients, and obligate recombinants were identified in two patients defining a candidate region of 17.7 cM between the centromeric marker D17S1863 (III-4) and the telomeric marker D17S1795 (III-6). Comparison of the linked alleles showed that in families DR2 and DR8 identical risk haplotypes were segregating for all STR markers within the region flanked by D17S1814 and D17S1795. This finding implies that families DR2 and DR8 are genetically related and originate from an unknown common founder. Therefore, by combining the segregation data of families DR2 and DR8, we were able to reduce the minimal candidate region to 8.04 cM between D17S1818 and D17S1795 delineated by haplotype sharing at the centromeric site and a recombinant in family DR2 (III-4) at the telomeric site. As the DR2 and DR8 families are genetically related, LOD scores of each family were summed, achieving a maximum LOD score of 5.28 at D17S931.


Pedigrees of Belgian FTLD families DR2 (A) and DR8 (B) showing chromosome 17q21 haplotypes of selected family members based on 18 informative STR markers; allele lengths are indicated in base pairs. The disease haplotype is boxed in black. Inferred haplotypes are shown between parentheses. For deceased patients, genotype data of at-risk offspring was used to deduce their haplotypes. For confidentiality reasons haplotypes are shown only for patients and obligate carriers; the number of at-risk individuals included in the genotyping is indicated within diamonds. In family DR8, the risk haplotype was arbitrarily set for I-1. An arrowhead indicates the index patient.
Segregation analysis of MAPT SNP16 in families DR2 and DR8 indicated that the shared disease haplotype contained the rare extended MAPT H2 haplotype. All patients were heterozygous H1/H2 except patient III-32 of family DR8 who was homozygous H2/H2.
DR2–DR8 haplotype sharing analysis in the Belgian FTLD patient series
Genotyping of 14 STR markers spanning the 8 cM DR2–DR8 disease locus in 98 Belgian FTLD patients revealed that the DR2–DR8 linked alleles and MAPT H2 haplotype were shared by five additional familial FTLD patients (DR25.1, DR26.1, DR27.1, DR28.1 and DR31.1; Table 3). Additional blood samples were collected in these FTLD families: DR25 (n = 6), DR26 (n = 2), DR27 (n = 8), DR28 (n = 4) and DR31 (n = 3). Segregation analysis in relatives of the five index patients confirmed that the shared alleles constituted a single haplotype, which co-segregated with the disease (Fig. 3). The DR2–DR8 haplotype was absent in 92 control chromosomes.
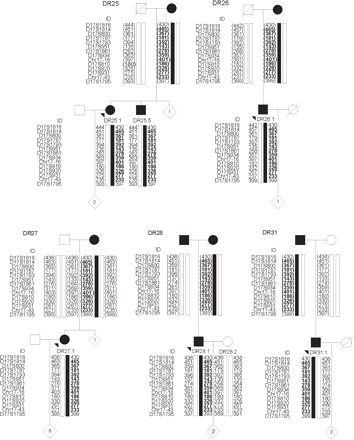
Pedigrees of Belgian FTLD families DR25, DR26, DR27, DR28 and DR31, showing chromosome 17q21 haplotypes of selected relatives based on 14 STR markers; allele lengths are indicated in base pairs. The index patients (indicated with an arrowhead) were selected on the basis of allele sharing with the DR2–DR8 ancestral haplotype. The disease haplotype is boxed in black. The DR2–DR8 ancestral haplotype is highlighted in bold. Inferred haplotypes are shown between parentheses. For confidentiality reasons haplotypes are shown only for patients and obligate carriers; the number of at-risk individuals included in the genotyping is indicated within diamonds.
Allele sharing analysis of STR markers and MAPT haplotypes spanning the 8 cM DR2–DR8 ancestral haplotype
| Marker | Linked allele (bp) in DR2–DR8a | Frequency of linked alleles (%)b | Patient of Belgian FTLD families | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR2: III-6 | DR8: III-32 | DR25.1 | DR26.1 | DR27.1 | DR28.1 | DR31.1 | |||||||||
| D17S1814 | 465 | 19 | 465-463 | 465-451 | 465-451 | 465-457 | 465-463 | 465-451 | 465-451 | ||||||
| D17S800 | 367 | 10 | 367-361 | 367-361 | 367-361 | 367-361 | 367-365 | 367-359 | 367-361 | ||||||
| D17S1787 | 181 | 35 | 181-179 | 181-179 | 181-177 | 181-179 | 181-177 | 181-181 | 181-177 | ||||||
| D17S1793 | 392 | 81 | 392-392 | 392-388 | 392-394 | 392-392 | 392-392 | 392-396 | 392-392 | ||||||
| D17S951 | 143 | 23 | 143-135 | 143-137 | 143-135 | 143-135 | 143-143 | 143-137 | 143-133 | ||||||
| D17S1861 | 278 | 6 | 278-264 | 278-280 | 278-262 | 278-268 | 278-276 | 278-274 | 278-274 | ||||||
| D17S934 | 359 | 27 | 359-363 | 359-359 | 359-363 | 359-357 | 359-371 | 359-361 | 359-359 | ||||||
| Chr17-16 | 401 | 22 | 401-397 | 401-397 | 401-397 | 401-397 | 401-403 | 401-397 | 401-401 | ||||||
| D17S810 | 186 | 30 | 186-182 | 186-186 | 186-182 | 186-182 | 186-180 | 186-180 | 186-180 | ||||||
| MAPT haplotypec | H2 | 33 | H2-H1 | H2-H2 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | ||||||
| D17S920 | 326 | 64 | 326-330 | 326-326 | 326-326 | 326-332 | 326-330 | 326-332 | 326-330 | ||||||
| D17S931 | 277 | 9 | 277-267 | 277-265 | 277-267 | 277-267 | 277-267 | 277-267 | 277-275 | ||||||
| Chr17-43 | 233 | 47 | 233-233 | 233-241 | 233-233 | 233-237 | 233-221 | 233-235 | 233-233 | ||||||
| Marker | Linked allele (bp) in DR2–DR8a | Frequency of linked alleles (%)b | Patient of Belgian FTLD families | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR2: III-6 | DR8: III-32 | DR25.1 | DR26.1 | DR27.1 | DR28.1 | DR31.1 | |||||||||
| D17S1814 | 465 | 19 | 465-463 | 465-451 | 465-451 | 465-457 | 465-463 | 465-451 | 465-451 | ||||||
| D17S800 | 367 | 10 | 367-361 | 367-361 | 367-361 | 367-361 | 367-365 | 367-359 | 367-361 | ||||||
| D17S1787 | 181 | 35 | 181-179 | 181-179 | 181-177 | 181-179 | 181-177 | 181-181 | 181-177 | ||||||
| D17S1793 | 392 | 81 | 392-392 | 392-388 | 392-394 | 392-392 | 392-392 | 392-396 | 392-392 | ||||||
| D17S951 | 143 | 23 | 143-135 | 143-137 | 143-135 | 143-135 | 143-143 | 143-137 | 143-133 | ||||||
| D17S1861 | 278 | 6 | 278-264 | 278-280 | 278-262 | 278-268 | 278-276 | 278-274 | 278-274 | ||||||
| D17S934 | 359 | 27 | 359-363 | 359-359 | 359-363 | 359-357 | 359-371 | 359-361 | 359-359 | ||||||
| Chr17-16 | 401 | 22 | 401-397 | 401-397 | 401-397 | 401-397 | 401-403 | 401-397 | 401-401 | ||||||
| D17S810 | 186 | 30 | 186-182 | 186-186 | 186-182 | 186-182 | 186-180 | 186-180 | 186-180 | ||||||
| MAPT haplotypec | H2 | 33 | H2-H1 | H2-H2 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | ||||||
| D17S920 | 326 | 64 | 326-330 | 326-326 | 326-326 | 326-332 | 326-330 | 326-332 | 326-330 | ||||||
| D17S931 | 277 | 9 | 277-267 | 277-265 | 277-267 | 277-267 | 277-267 | 277-267 | 277-275 | ||||||
| Chr17-43 | 233 | 47 | 233-233 | 233-241 | 233-233 | 233-237 | 233-221 | 233-235 | 233-233 | ||||||
Linked alleles are in bold; bAllele frequencies were calculated in 92 control chromosomes; cMAPT haplotypes were determined by genotyping the MAPT htSNP16 according to Rademakers et al., 2005.
Allele sharing analysis of STR markers and MAPT haplotypes spanning the 8 cM DR2–DR8 ancestral haplotype
| Marker | Linked allele (bp) in DR2–DR8a | Frequency of linked alleles (%)b | Patient of Belgian FTLD families | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR2: III-6 | DR8: III-32 | DR25.1 | DR26.1 | DR27.1 | DR28.1 | DR31.1 | |||||||||
| D17S1814 | 465 | 19 | 465-463 | 465-451 | 465-451 | 465-457 | 465-463 | 465-451 | 465-451 | ||||||
| D17S800 | 367 | 10 | 367-361 | 367-361 | 367-361 | 367-361 | 367-365 | 367-359 | 367-361 | ||||||
| D17S1787 | 181 | 35 | 181-179 | 181-179 | 181-177 | 181-179 | 181-177 | 181-181 | 181-177 | ||||||
| D17S1793 | 392 | 81 | 392-392 | 392-388 | 392-394 | 392-392 | 392-392 | 392-396 | 392-392 | ||||||
| D17S951 | 143 | 23 | 143-135 | 143-137 | 143-135 | 143-135 | 143-143 | 143-137 | 143-133 | ||||||
| D17S1861 | 278 | 6 | 278-264 | 278-280 | 278-262 | 278-268 | 278-276 | 278-274 | 278-274 | ||||||
| D17S934 | 359 | 27 | 359-363 | 359-359 | 359-363 | 359-357 | 359-371 | 359-361 | 359-359 | ||||||
| Chr17-16 | 401 | 22 | 401-397 | 401-397 | 401-397 | 401-397 | 401-403 | 401-397 | 401-401 | ||||||
| D17S810 | 186 | 30 | 186-182 | 186-186 | 186-182 | 186-182 | 186-180 | 186-180 | 186-180 | ||||||
| MAPT haplotypec | H2 | 33 | H2-H1 | H2-H2 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | ||||||
| D17S920 | 326 | 64 | 326-330 | 326-326 | 326-326 | 326-332 | 326-330 | 326-332 | 326-330 | ||||||
| D17S931 | 277 | 9 | 277-267 | 277-265 | 277-267 | 277-267 | 277-267 | 277-267 | 277-275 | ||||||
| Chr17-43 | 233 | 47 | 233-233 | 233-241 | 233-233 | 233-237 | 233-221 | 233-235 | 233-233 | ||||||
| Marker | Linked allele (bp) in DR2–DR8a | Frequency of linked alleles (%)b | Patient of Belgian FTLD families | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR2: III-6 | DR8: III-32 | DR25.1 | DR26.1 | DR27.1 | DR28.1 | DR31.1 | |||||||||
| D17S1814 | 465 | 19 | 465-463 | 465-451 | 465-451 | 465-457 | 465-463 | 465-451 | 465-451 | ||||||
| D17S800 | 367 | 10 | 367-361 | 367-361 | 367-361 | 367-361 | 367-365 | 367-359 | 367-361 | ||||||
| D17S1787 | 181 | 35 | 181-179 | 181-179 | 181-177 | 181-179 | 181-177 | 181-181 | 181-177 | ||||||
| D17S1793 | 392 | 81 | 392-392 | 392-388 | 392-394 | 392-392 | 392-392 | 392-396 | 392-392 | ||||||
| D17S951 | 143 | 23 | 143-135 | 143-137 | 143-135 | 143-135 | 143-143 | 143-137 | 143-133 | ||||||
| D17S1861 | 278 | 6 | 278-264 | 278-280 | 278-262 | 278-268 | 278-276 | 278-274 | 278-274 | ||||||
| D17S934 | 359 | 27 | 359-363 | 359-359 | 359-363 | 359-357 | 359-371 | 359-361 | 359-359 | ||||||
| Chr17-16 | 401 | 22 | 401-397 | 401-397 | 401-397 | 401-397 | 401-403 | 401-397 | 401-401 | ||||||
| D17S810 | 186 | 30 | 186-182 | 186-186 | 186-182 | 186-182 | 186-180 | 186-180 | 186-180 | ||||||
| MAPT haplotypec | H2 | 33 | H2-H1 | H2-H2 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | H2-H1 | ||||||
| D17S920 | 326 | 64 | 326-330 | 326-326 | 326-326 | 326-332 | 326-330 | 326-332 | 326-330 | ||||||
| D17S931 | 277 | 9 | 277-267 | 277-265 | 277-267 | 277-267 | 277-267 | 277-267 | 277-275 | ||||||
| Chr17-43 | 233 | 47 | 233-233 | 233-241 | 233-233 | 233-237 | 233-221 | 233-235 | 233-233 | ||||||
Linked alleles are in bold; bAllele frequencies were calculated in 92 control chromosomes; cMAPT haplotypes were determined by genotyping the MAPT htSNP16 according to Rademakers et al., 2005.
Clinical features associated with the DR2–DR8 haplotype
When considering all patients with the DR2–DR8 haplotype, we calculated a mean onset age of 63.25 years (n = 28; range 51–75 years) and a mean disease duration 6.05 years (n = 20; range 1–20 years). More detailed clinical characteristics of 11 DR2–DR8 haplotype carriers are compared in Table 4. In 9 out of 11 FTLD patients, language impairments ranging from progressive non-fluent aphasia (PNFA) (n =4) to reduced spontaneous speech (n = 3), word-finding problems (n = 1) or post-stroke aphasia (n = 1) were present at presentation. In addition and consistent with prominent phatic symptoms, neuroimaging revealed lateralization of the brain atrophy and/or hypoperfusion to the left side in the majority (7 out of 11) of the patients (Table 4; Fig. 4). In the one patient with progressive behavioural and personality changes but without speech impairment (DR27.1), the structural and functional defect was more pronounced at the right side.
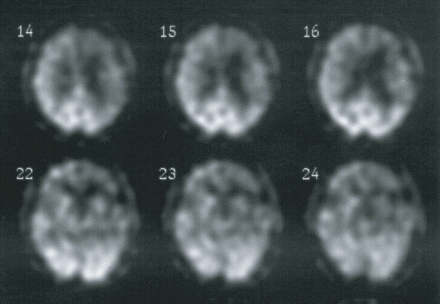
Brain perfusion single photon emission computed tomography (SPECT) of FTLD patient DR31.1 four years after onset of the disease showing relative bilateral frontal and temporal hypoperfusion, with left more pronounced than right.
Clinical findings in the FTDL DR2–DR8 haplotype carriers
| Individual | Gender | Onset age (years) | Age at death (†) or current age (*) (years) | Disease duration (years) | Presenting symptoms | Presenting diagnosis | Diagnosis at follow-up | Structural neuroimaging (CT/MRI) | Functional neuroimaging (PET/SPECT) |
|---|---|---|---|---|---|---|---|---|---|
| DR2 III-8 | M | 66 | 71 (†) | 5 | Impaired memory and concentration | FTD | FTD | Global mainly subcortical atrophy (CT) | NA |
| DR2 III-10 | M | 69 | 71 (*) | >2 | Apathy, reduced spontaneous speech, agrammatism, verbal perseverations, dysarthria | FTD | NA | Global moderate cortical and subcortical atrophy (MRI) | Relative frontal and frontoparietal HP, left > right. Relative HP of the left thalamus. Diastasis of frontal cortical activity (SPECT) |
| DR2 III-6 | F | 63 | 71 (*) | >8 | Non-fluent aphasia, personality changes (mainly apathy) | PNFA | FTD | Global cortical and subcortical atrophy, right > left; PWML (MRI) | Relative bilateral frontal, parietal and temporal HP, right > left (SPECT) |
| DR8 III-28 | F | 62 | 68 (*) | >6 | Personality changes (apathy), behavioural disturbances (psychosis, disinhibition), word-finding difficulties and impaired memory | FTD | FTD | Frontotemporoparietal cortical and subcortical atrophy, left > right (MRI) | Relative bilateral frontal HP, left > right (SPECT) |
| DR8 III-18 | F | 51 | 55 (†) | 4 | Impaired memory, reduced spontaneous speech, echolalia, apathy | FTD | FTD | Global cortical and subcortical atrophy (CT) | Severe relative bifrontal HP, left > right (SPECT) |
| DR31.1 | M | 66 | 70 (†) | 4 | Non-fluent aphasia | PNFA | FTD | Global cortical and minor subcortical temporal atrophy, left > right (MRI) | Marked relative bilateral frontal and temporal HP, left > right Diastasis of frontal cortical activity (SPECT) |
| DR26.1 | M | 65 | 68 (†) | 3 | Progressive apraxia of speech | PNFA | FTD | Global subcortical and cortical atrophy, maximal frontally and temporally, left > right (MRI) | Relative frontal, temporal and parietal HP, left > right. Relative HP of the basal ganglia and lentiform nucleus. Right cerebellar HP (SPECT) |
| DR25.1 | F | 69 | 75 (*) | >3 | Behavioural disturbances, personality changes, reduced spontaneous speech | FTD | FTD | Cortical and subcortical frontal atrophy; PWML (CT) | Severe relative bilateral frontal, parietal and temporal HP Scintigraphic indications of subcortical loss (SPECT) |
| DR25.5 | M | 70 | 71 (*) | >1 | Behavioural disturbances and personality changes, worsening (of pre-existing post-stroke) aphasia | FTD | NA | Cortical and subcortical atrophy, maximal frontally, left > right; PWML (MRI) | Bilateral frontal, parietal and temporal HP, left > right Right cerebellar HP (PET) |
| DR27.1 | F | 58 | 64 (†) | 6 | Behavioural disturbances, personality changes | FTD | FTD | Cortical and subcortical atrophy, maximal frontotemporally, right > left; PWML (MRI) | Bilateral frontal, temporal and parietal HP, right > left. Right HP at parieto-occipital transition Left cerebellar HP (PET) |
| DR28.1 | M | 57 | 62 | >5 | Non-fluent aphasia | PNFA | FTD | NA | Relative frontal, temporal and parietal HP, left > right (SPECT) |
| Individual | Gender | Onset age (years) | Age at death (†) or current age (*) (years) | Disease duration (years) | Presenting symptoms | Presenting diagnosis | Diagnosis at follow-up | Structural neuroimaging (CT/MRI) | Functional neuroimaging (PET/SPECT) |
|---|---|---|---|---|---|---|---|---|---|
| DR2 III-8 | M | 66 | 71 (†) | 5 | Impaired memory and concentration | FTD | FTD | Global mainly subcortical atrophy (CT) | NA |
| DR2 III-10 | M | 69 | 71 (*) | >2 | Apathy, reduced spontaneous speech, agrammatism, verbal perseverations, dysarthria | FTD | NA | Global moderate cortical and subcortical atrophy (MRI) | Relative frontal and frontoparietal HP, left > right. Relative HP of the left thalamus. Diastasis of frontal cortical activity (SPECT) |
| DR2 III-6 | F | 63 | 71 (*) | >8 | Non-fluent aphasia, personality changes (mainly apathy) | PNFA | FTD | Global cortical and subcortical atrophy, right > left; PWML (MRI) | Relative bilateral frontal, parietal and temporal HP, right > left (SPECT) |
| DR8 III-28 | F | 62 | 68 (*) | >6 | Personality changes (apathy), behavioural disturbances (psychosis, disinhibition), word-finding difficulties and impaired memory | FTD | FTD | Frontotemporoparietal cortical and subcortical atrophy, left > right (MRI) | Relative bilateral frontal HP, left > right (SPECT) |
| DR8 III-18 | F | 51 | 55 (†) | 4 | Impaired memory, reduced spontaneous speech, echolalia, apathy | FTD | FTD | Global cortical and subcortical atrophy (CT) | Severe relative bifrontal HP, left > right (SPECT) |
| DR31.1 | M | 66 | 70 (†) | 4 | Non-fluent aphasia | PNFA | FTD | Global cortical and minor subcortical temporal atrophy, left > right (MRI) | Marked relative bilateral frontal and temporal HP, left > right Diastasis of frontal cortical activity (SPECT) |
| DR26.1 | M | 65 | 68 (†) | 3 | Progressive apraxia of speech | PNFA | FTD | Global subcortical and cortical atrophy, maximal frontally and temporally, left > right (MRI) | Relative frontal, temporal and parietal HP, left > right. Relative HP of the basal ganglia and lentiform nucleus. Right cerebellar HP (SPECT) |
| DR25.1 | F | 69 | 75 (*) | >3 | Behavioural disturbances, personality changes, reduced spontaneous speech | FTD | FTD | Cortical and subcortical frontal atrophy; PWML (CT) | Severe relative bilateral frontal, parietal and temporal HP Scintigraphic indications of subcortical loss (SPECT) |
| DR25.5 | M | 70 | 71 (*) | >1 | Behavioural disturbances and personality changes, worsening (of pre-existing post-stroke) aphasia | FTD | NA | Cortical and subcortical atrophy, maximal frontally, left > right; PWML (MRI) | Bilateral frontal, parietal and temporal HP, left > right Right cerebellar HP (PET) |
| DR27.1 | F | 58 | 64 (†) | 6 | Behavioural disturbances, personality changes | FTD | FTD | Cortical and subcortical atrophy, maximal frontotemporally, right > left; PWML (MRI) | Bilateral frontal, temporal and parietal HP, right > left. Right HP at parieto-occipital transition Left cerebellar HP (PET) |
| DR28.1 | M | 57 | 62 | >5 | Non-fluent aphasia | PNFA | FTD | NA | Relative frontal, temporal and parietal HP, left > right (SPECT) |
FTLD subdiagnoses of progressive non-fluent aphasia (PNFA) and frontotemporal dementia (FTD) were made according to established criteria (Neary et al., 1998). Neuropsychological assessment and functional neuroimaging (single photon emission computed tomography, SPECT or positron emission tomography, PET) were used to further support the clinical diagnosis of FTLD (Pickut et al., 1997). HP = hypoperfusion, NA = not available, PWML = periventricular white matter lesions; MRI = magnetic resonance imaging.
Clinical findings in the FTDL DR2–DR8 haplotype carriers
| Individual | Gender | Onset age (years) | Age at death (†) or current age (*) (years) | Disease duration (years) | Presenting symptoms | Presenting diagnosis | Diagnosis at follow-up | Structural neuroimaging (CT/MRI) | Functional neuroimaging (PET/SPECT) |
|---|---|---|---|---|---|---|---|---|---|
| DR2 III-8 | M | 66 | 71 (†) | 5 | Impaired memory and concentration | FTD | FTD | Global mainly subcortical atrophy (CT) | NA |
| DR2 III-10 | M | 69 | 71 (*) | >2 | Apathy, reduced spontaneous speech, agrammatism, verbal perseverations, dysarthria | FTD | NA | Global moderate cortical and subcortical atrophy (MRI) | Relative frontal and frontoparietal HP, left > right. Relative HP of the left thalamus. Diastasis of frontal cortical activity (SPECT) |
| DR2 III-6 | F | 63 | 71 (*) | >8 | Non-fluent aphasia, personality changes (mainly apathy) | PNFA | FTD | Global cortical and subcortical atrophy, right > left; PWML (MRI) | Relative bilateral frontal, parietal and temporal HP, right > left (SPECT) |
| DR8 III-28 | F | 62 | 68 (*) | >6 | Personality changes (apathy), behavioural disturbances (psychosis, disinhibition), word-finding difficulties and impaired memory | FTD | FTD | Frontotemporoparietal cortical and subcortical atrophy, left > right (MRI) | Relative bilateral frontal HP, left > right (SPECT) |
| DR8 III-18 | F | 51 | 55 (†) | 4 | Impaired memory, reduced spontaneous speech, echolalia, apathy | FTD | FTD | Global cortical and subcortical atrophy (CT) | Severe relative bifrontal HP, left > right (SPECT) |
| DR31.1 | M | 66 | 70 (†) | 4 | Non-fluent aphasia | PNFA | FTD | Global cortical and minor subcortical temporal atrophy, left > right (MRI) | Marked relative bilateral frontal and temporal HP, left > right Diastasis of frontal cortical activity (SPECT) |
| DR26.1 | M | 65 | 68 (†) | 3 | Progressive apraxia of speech | PNFA | FTD | Global subcortical and cortical atrophy, maximal frontally and temporally, left > right (MRI) | Relative frontal, temporal and parietal HP, left > right. Relative HP of the basal ganglia and lentiform nucleus. Right cerebellar HP (SPECT) |
| DR25.1 | F | 69 | 75 (*) | >3 | Behavioural disturbances, personality changes, reduced spontaneous speech | FTD | FTD | Cortical and subcortical frontal atrophy; PWML (CT) | Severe relative bilateral frontal, parietal and temporal HP Scintigraphic indications of subcortical loss (SPECT) |
| DR25.5 | M | 70 | 71 (*) | >1 | Behavioural disturbances and personality changes, worsening (of pre-existing post-stroke) aphasia | FTD | NA | Cortical and subcortical atrophy, maximal frontally, left > right; PWML (MRI) | Bilateral frontal, parietal and temporal HP, left > right Right cerebellar HP (PET) |
| DR27.1 | F | 58 | 64 (†) | 6 | Behavioural disturbances, personality changes | FTD | FTD | Cortical and subcortical atrophy, maximal frontotemporally, right > left; PWML (MRI) | Bilateral frontal, temporal and parietal HP, right > left. Right HP at parieto-occipital transition Left cerebellar HP (PET) |
| DR28.1 | M | 57 | 62 | >5 | Non-fluent aphasia | PNFA | FTD | NA | Relative frontal, temporal and parietal HP, left > right (SPECT) |
| Individual | Gender | Onset age (years) | Age at death (†) or current age (*) (years) | Disease duration (years) | Presenting symptoms | Presenting diagnosis | Diagnosis at follow-up | Structural neuroimaging (CT/MRI) | Functional neuroimaging (PET/SPECT) |
|---|---|---|---|---|---|---|---|---|---|
| DR2 III-8 | M | 66 | 71 (†) | 5 | Impaired memory and concentration | FTD | FTD | Global mainly subcortical atrophy (CT) | NA |
| DR2 III-10 | M | 69 | 71 (*) | >2 | Apathy, reduced spontaneous speech, agrammatism, verbal perseverations, dysarthria | FTD | NA | Global moderate cortical and subcortical atrophy (MRI) | Relative frontal and frontoparietal HP, left > right. Relative HP of the left thalamus. Diastasis of frontal cortical activity (SPECT) |
| DR2 III-6 | F | 63 | 71 (*) | >8 | Non-fluent aphasia, personality changes (mainly apathy) | PNFA | FTD | Global cortical and subcortical atrophy, right > left; PWML (MRI) | Relative bilateral frontal, parietal and temporal HP, right > left (SPECT) |
| DR8 III-28 | F | 62 | 68 (*) | >6 | Personality changes (apathy), behavioural disturbances (psychosis, disinhibition), word-finding difficulties and impaired memory | FTD | FTD | Frontotemporoparietal cortical and subcortical atrophy, left > right (MRI) | Relative bilateral frontal HP, left > right (SPECT) |
| DR8 III-18 | F | 51 | 55 (†) | 4 | Impaired memory, reduced spontaneous speech, echolalia, apathy | FTD | FTD | Global cortical and subcortical atrophy (CT) | Severe relative bifrontal HP, left > right (SPECT) |
| DR31.1 | M | 66 | 70 (†) | 4 | Non-fluent aphasia | PNFA | FTD | Global cortical and minor subcortical temporal atrophy, left > right (MRI) | Marked relative bilateral frontal and temporal HP, left > right Diastasis of frontal cortical activity (SPECT) |
| DR26.1 | M | 65 | 68 (†) | 3 | Progressive apraxia of speech | PNFA | FTD | Global subcortical and cortical atrophy, maximal frontally and temporally, left > right (MRI) | Relative frontal, temporal and parietal HP, left > right. Relative HP of the basal ganglia and lentiform nucleus. Right cerebellar HP (SPECT) |
| DR25.1 | F | 69 | 75 (*) | >3 | Behavioural disturbances, personality changes, reduced spontaneous speech | FTD | FTD | Cortical and subcortical frontal atrophy; PWML (CT) | Severe relative bilateral frontal, parietal and temporal HP Scintigraphic indications of subcortical loss (SPECT) |
| DR25.5 | M | 70 | 71 (*) | >1 | Behavioural disturbances and personality changes, worsening (of pre-existing post-stroke) aphasia | FTD | NA | Cortical and subcortical atrophy, maximal frontally, left > right; PWML (MRI) | Bilateral frontal, parietal and temporal HP, left > right Right cerebellar HP (PET) |
| DR27.1 | F | 58 | 64 (†) | 6 | Behavioural disturbances, personality changes | FTD | FTD | Cortical and subcortical atrophy, maximal frontotemporally, right > left; PWML (MRI) | Bilateral frontal, temporal and parietal HP, right > left. Right HP at parieto-occipital transition Left cerebellar HP (PET) |
| DR28.1 | M | 57 | 62 | >5 | Non-fluent aphasia | PNFA | FTD | NA | Relative frontal, temporal and parietal HP, left > right (SPECT) |
FTLD subdiagnoses of progressive non-fluent aphasia (PNFA) and frontotemporal dementia (FTD) were made according to established criteria (Neary et al., 1998). Neuropsychological assessment and functional neuroimaging (single photon emission computed tomography, SPECT or positron emission tomography, PET) were used to further support the clinical diagnosis of FTLD (Pickut et al., 1997). HP = hypoperfusion, NA = not available, PWML = periventricular white matter lesions; MRI = magnetic resonance imaging.
Neuropathology of DR2–DR8 haplotype carrier DR31.1
On macroscopic examination, the brain of the index patient of family DR31 (DR31.1) demonstrated severe frontotemporal atrophy mainly affecting the frontal gyri. Coronal frozen sections of the frontal regions confirmed a clear neuronal loss, severe demyelination of the white matter, fibrillary gliosis, microspongiosis and an enormous dilatation of the frontal horn of the lateral ventricle. In addition, PAS staining revealed numerous lipofuscin granules in neurons, astrocytes and pericapillary pericytes as well as numerous subpial and perivascular corpora amylacea. Sections stained with cresyl violet showed severe neuronal loss, astrocytic gliosis and microspongiosis. No ballooned or chromatolytic neurons were observed. In general, the lesions were most outspoken in the prefrontal regions and much less prominent in temporal regions. Immunohistochemistry was performed on paraffin sections from select regions: superior frontal gyrus, cingular gyrus, superior temporal gyrus, hippocampus, parahippocampal gyrus, occipital gyrus, cerebellum, substantia nigra and pons. With exception of the right hippocampus where a few AT8-positive neurofibrillary tangles were observed, all other brain regions were AT8-negative. Staining with 4G8 revealed rare perivascular Aβ deposits in the entorhinal zone of the hippocampus but was completely negative in all other regions. With an antibody directed against ubiquitin, rare intraneuronal cytoplasmic structures were observed in the superior frontal gyrus, superior temporal gyrus and hippocampus. These perinuclear inclusions had a pleiomorphic appearance ranging from thin filamentous threads to more tortuous and granular structures (Fig. 5A and B). No Lewy bodies were observed. Very rare ubiquitin-positive intranuclear inclusions were observed exclusively in sections from the superior frontal gyrus (Fig. 5A). These intranuclear ubiquitin-positive structures were morphologically identical to the ‘cat eye’-like inclusions described previously in inherited tau-negative FTLD brains (Rosso et al., 2001; Rademakers et al., 2002). The neuropathological diagnosis was consistent with FTDU.
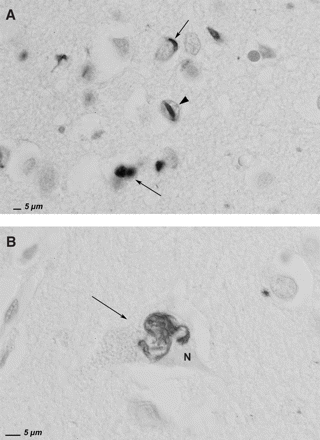
Ubiquitin immunohistochemistry of superior frontal gyrus of FTLD patient DR31.1. (A) The arrowhead points towards an ubiquitin-positive cat eye-shaped intranuclear inclusion. The arrows show ubiquitin-positive structures in neuronal perikarya. (B) The arrow shows a filamentous, tortuous ubiquitin-positive structure in the perikaryon of a neuron, the nucleus being indicated by the letter N. (Scale bar = 5 μm).
Discussion
In this study we examined the frequency of tau-negative FTLD linked to 17q21 in a series of 98 well-characterized Belgian FTLD patients. The male : female ratio and onset ages in the present Belgian FTLD patient sample are in line with previously reported FTLD series (Johnson et al., 2005). Also, the percentage of familial FTLD (43%) was very similar to that reported in a nationwide study on the prevalence of FTLD conducted in The Netherlands (Rosso et al., 2003) and other studies (Poorkaj et al., 2001a). Although the number of autopsies is small (n = 7) to draw conclusions on the prevalence of the different pathological subtypes, tau-negative FTLD (FTDU and DLDH) was clearly much more prevalent (5 out of 7; 71%) than tau-positive FTLD (1 out of 7; 29%), in line with previous reports (Mann et al., 2000; Morris et al., 2001; Rosso et al., 2003; Hodges et al., 2004; Johnson et al., 2005). Surprisingly, however, the MAPT mutation frequency (1%) was low compared with other studies (5–20%) (Rizzu et al., 1999; Poorkaj et al., 2001a) and suggested that (an)other genetic defect(s), different from classical MAPT mutations, explain(s) the majority of familial FTLD in Belgium.
From two patients of our Belgian FTLD population, we collected two multiplex families, DR2 and DR8. Both families were linked to the same 17q21 haplotype, indicating that they are part of a single extended pedigree giving a summed LOD score of 5.28 at D17S931. The DR2–DR8 candidate region of 8 cM encompassed the minimal 4.8 cM candidate region we previously identified in the Dutch 17q21-linked FTDU family 1083 (Rademakers et al., 2002). Comparing haplotype data of the Belgian FTDU families DR2–DR8 with that obtained in Dutch family 1083 (Rademakers et al., 2002) did not identify significant allele sharing pointing to allelic heterogeneity at this locus. Also, in the Belgian FTDU families the linked haplotype comprised the extended MAPT haplotype H2, whereas other FTDU families linked to 17q21 segregated the extended MAPT haplotype H1 as part of their disease haplotype (1083, Dutch III). The latter observation argues against a causal role of the H1–H2 inversion polymorphism in 17q21-linked tau-negative FTLD. However, since in all these families MAPT is comprised within the disease haplotype, we cannot exclude that MAPT does play a role in the disease mechanism of tau-negative FTLD although different from that in tauopathies with aggregated tau and MAPT mutations.
Interestingly, we identified the DR2–DR8 haplotype in another five patients with positive family history in the same FTLD series. Together with the two index patients from families DR2 and DR8, the DR2–DR8 haplotype therefore explained 7% (7 out of 98) of FTLD in the overall series and 17% (7 out of 42) of familial FTLD. All DR2–DR8 haplotype carriers were living throughout the region of the neighbouring provinces of Antwerp and Flemish Brabant, in the Dutch-speaking region of Belgium, Flanders. The sharing of a common haplotype and the close geographical proximity of the seven families are indicative of a common ancestor. Most probably our data underestimated the actual frequency of the underlying genetic defect since in our FTLD series we observed several patients who shared alleles in smaller segments of the DR2–DR8 ancestral haplotype. Information of these partial haplotypes would be most helpful in reducing the candidate region to a more amenable size for gene cloning. However, several of the partial haplotypes were also present in control individuals and thus potentially unrelated to disease. We are currently collecting additional family members of each potential partial sharer for haplotype segregation studies to allow identification of true sharers.
Clinically, DR2–DR8 haplotype carriers presented with FTLD at a mean onset age of 63.25 years and had disease duration of 6.05 years. These clinical features are similar to what is reported for the other conclusive 17q21-linked tau-negative families 1083, HDDD2 and Dutch III (Rademakers et al., 2002). Interestingly, the DR2–DR8 haplotype carriers often presented with language impairments, which correlated with predominant structural and/or functional brain defects at the left side. Disproportionate dysphasia was also reported in family HDDD2 (Lendon et al., 1998) but not in the Dutch families 1083 (Rademakers et al., 2002) and Dutch III (Rosso et al., 2001). These results suggest that the presence of phatic impairments might define an allelic subtype of 17q21-linked tau-negative FTLD.
We have not yet obtained brain pathology in the conclusively linked pedigree DR8 (LOD score > 3), only in one of the ancestral haplotype carriers (DR31.1). In this patient, the ubiquitin positive cat eye-shaped intranuclear inclusions were rare but morphologically identical to those described previously in other tau-negative 17q21-linked FTLD families 1083 and Dutch III (Rosso et al., 2001; Rademakers et al., 2002). In contrast, in the HDDD2 family, DLDH was the pathological diagnosis (Lendon et al., 1998; Zhukareva et al., 2001). Given the rarity of such inclusions presented here, we speculate that FTDU and DLDH belong to a spectrum of pathological manifestations that can be caused by the same or a similar genetic defect at 17q21. We do, however, realize that extrapolating the FTDU pathology data to the 17q21-linked families might seem premature. However, the sharing data we observed is highly significant, since the ancestral haplotype was absent from 92 control chromosomes, supporting our interpretation that the sharing between the seven families is unlikely coincidental and that they are part of the same founder pedigree. Further, we have already obtained informed consent for brain autopsy of several patients in the seven founder families in case of death, which will allow future detailed studies of brain pathology and FTDU specifically. Also, availability of frozen brain material would allow studies of MAPT mRNA and protein stability. Previous such studies in the Dutch III FTDU family showed normal MAPT mRNA and isoform distributions (Rosso et al., 2001). This is in contrast with the report of loss of all tau protein isoforms in family HDDD2 but normal tau mRNA (Zhukareva et al., 2001).
In conclusion, we identified a highly penetrant MAPT containing ancestral haplotype in a population of 98 genealogically unrelated Belgian FTLD patients explaining a substantial portion of familial FTLD (17%). The haplotype carriers were characterized by frequent language impairments, the neuropathological presence of tau-negative ubiquitin-positive neuronal inclusions and absence of MAPT mutations. In contrast, in the same patient series we have identified only one MAPT mutation carrier among the familial FTLD patients (2%), indicating that 17q21-linked tau-negative FTLD is much more frequent among Belgian FTLD patients. Our findings strongly suggest that the ancestral DR2–DR8 haplotype harbours a frequent causal mutation for 17q21-linked tau-negative FTLD.
Electronic database information
Accession numbers and URLs for data presented in this article are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
AD&FTD Mutation Database, http://www.molgen.ua.ac.be/FTDMutations/
Marshfield Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/
VIB8 Genetic Service Facility, http://www.vibgeneticservicefacility.be
UCSC Genome Bioinformatics, http://genome.ucsc.edu/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ [for microsatellite markers Chr17-16 (accession number AC008105.32), Chr17-19 (accession number AC091628.2), Chr17-43 (accession number AC068234.8) and for MAPT (accession number AC091628.2)].
The authors are grateful to the family members for their kind cooperation in this study, and to the personnel of the VIB8 Genetic Service Facility (http://www.vibgeneticservicefacility.be) for the genetic analyses. The research described in this paper was supported by the Special Research Fund of the University of Antwerp, the Fund for Scientific Research Flanders (FWO-F), the Interuniversity Attraction Poles program P5/19 of the Belgian Science Policy Office, the International Alzheimer Research Foundation, Belgium; the EU contract LSHM-CT-2003-503330 (APOPIS) and the Alzheimer's Association, USA. S.E., R.R. and M.C. are postdoctoral fellows and I.G. and V.B. are PhD fellows of the FWO-F. R.V. is a clinical investigator of the FWO-F. Funding to pay the Open Access publication charges for this article was provided by The Special Research Fund of the University of Antwerp.
References
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy.
Benson G. Tandem repeats finder: a program to analyze DNA sequences.
Brown J, Ashworth A, Gydesen S, Sorensen A, Rossor M, Hardy J, et al. Familial non-specific dementia maps to chromosome 3.
Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia.
Cruts M, Rademakers R, Gijselinck I, van der Zee J, Dermaut B, De Pooter T, et al. Genomic architecture of human 17q21 linked to frontotemporal dementia uncovers a highly homologous family of low copy repeats in the tau region.
Dermaut B, Kumar-Singh S, Engelborghs S, Theuns J, Rademakers R, Sacrens J, et al. A novel presenilin 1 mutation associated with Pick's disease but not beta-amyloid plaques.
Engelborghs S, Dermaut B, Goeman J, Saerens J, Marien P, Pickut BA, et al. Prospective Belgian study of neurodegenerative and vascular dementia: APOE genotype effects.
Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome
Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years.
Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia.
Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17.
Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients.
Kumar-Singh S, Cras P, Wang R, Kros JM, van Swieten J, Lubke U, et al. Dense-core senile plaques in the Flemish variant of Alzheimer's disease are vasocentric.
Lathrop GM, Lalouel JM, Julier C, Ott J. Multilocus linkage analysis in humans: detection of linkage and estimation of recombination.
Lendon CL, Lynch T, Norton J, Mckeel DW, Busfield F, Craddock N, et al. Hereditary dysphasic disinhibition dementia—a frontotemporal dementia linked to 17q21-22.
Mann DM, McDonagh AM, Snowden J, Neary D, Pickering-Brown SM. Molecular classification of the dementias.
Morris HR, Khan MN, Janssen JC, Brown JM, Perez-Tur J, Baker, M et al. The genetic and pathological classification of familial frontotemporal dementia.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria.
Ott A, Breteler MM, Van Harskamp F, Claus JJ, van der Cammen TJ, Grobbee DE, et al. Prevalence of Alzheimer's disease and vascular dementia: association with education. The Rotterdam study.
Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, et al. Alpha-synuclein promoter confers susceptibility to Parkinson's disease.
Pickut BA, Saerens J, Marien P, Borggreve F, Goeman J, Vandevivere J, et al. Discriminative use of SPECT in frontal lobe-type dementia versus (senile) dementia of the Alzheimer's type.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia.
Poorkaj P, Grossman M, Steinbart E, Payami H, Sadovnick A, Nochlin D, et al. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia.
Poorkaj P, Kas A, D'Souza I, Zhou Y, Pham Q, Stone M, et al. A genomic sequence analysis of the mouse and human microtubule-associated protein tau.
Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van den Broeck M, et al. Tau negative frontal lobe dementia at 17q
Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies.
Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy.
Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia.
Rizzu P, van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands.
Rosso SM, Kaat LD, Baks T, Joosse M, de Koning I, Pijnenburg Y, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study.
Rosso SM, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Niermeijer MF, et al. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q2l-22.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia.
Stankiewicz P, Lupski JR. Molecular-evolutionary mechanisms for genomic disorders.
Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J et al. A common inversion under selection in Europeans.
Stevens M, van Duijn CM, Kamphorst W, de Knijff P, Heutink P, van Gool WA, et al. Familial aggregation in frontotemporal dementia.
Author notes
1Neurodegenerative Brain Diseases Group, Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, 2Laboratory of Neurochemistry and Behavior, 3Laboratory of Neuropathology, Institute Born-Bunge, University of Antwerp, 4Department of Neurology, Ghent University Hospital, University of Ghent, 5Department of Neurology, University Hospital Gasthuisberg, Catholic University of Leuven, 6Department of Neurology and Memory Clinic, Middelheim General Hospital, Antwerp and 7Department of Neurology, OLV Hospital Aalst, Belgium


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}