




Abstract
Alzheimer's disease is an age-related progressive neurodegenerative disorder with an enormous unmet medical need. It is the most common form of dementia affecting ∼5% of adults over 65 years. In view of our ageing society the number of patients, as well as the economical and social impact, is expected to grow dramatically in the future. Currently available medications appear to be able to produce moderate symptomatic benefits but not to stop disease progression. The search for novel therapeutic approaches targeting the presumed underlying pathogenic mechanisms has been a major focus of research and it is expected that novel medications with disease-modifying properties will emerge from these efforts in the future. In this review, currently available drugs as well as novel therapeutic strategies, in particular those targeting amyloid and tau pathologies, are discussed.
Abbreviations
- Aβ
amyloid-β peptide
- AchE
acetylcholinesterase
- APP
amyloid precursor protein
- NFTs
neurofibrillary tangles
- NMDA
N-methyl-d-aspartate
- PHFs
paired helical filaments
- PS1 and PS2
presenilin-1 and -2
Introduction
Alzheimer's disease is the most common cause of progressive dementia in the elderly population. It is a chronic neurodegenerative disorder that leads to progressive disturbances of cognitive functions including memory, judgement, decision-making, orientation to physical surroundings and language (Nussbaum and Ellis, 2003). Characteristic neuropathological findings include selective neuronal and synaptic losses (Morrison and Hof, 1997), extracellular neuritic plaques containing the β-amyloid peptide (Glenner and Wong, 1984; Masters et al., 1985) and neurofibrillary tangles (NFTs) composed of hyperphosphorylated forms of the tau protein (Delacourte and Defossez, 1986; Grundke-Iqbal et al., 1986a, b; Kosik et al., 1986; Goedert et al., 1988, 1992; Wischik et al., 1988; Flament et al., 1989; Lee et al., 1991; Hasegawa et al., 1992; Sergeant et al., 1995). The clinical picture of dementia, as well as the histological findings of amyloid plaques and NFTs, was described as early as 1906 by the German psychiatrist Alois Alzheimer at a conference in Tübingen (reviewed by Maurer et al., 1997). His findings were published in his famous report ‘Über eine eigenartige Erkrankung der Hirnrinde’ [‘A characteristic disease of the cerebral cortex’] in 1907 (Alzheimer, 1907). In his 1911 publication, Alzheimer reported his second case of dementia and also included drawings of the typical neurofibrillary changes from his first case (Alzheimer, 1911; for reviews on Alzheimer's work and contributions of others in this context, seeBick, 1994; Maurer et al., 1997; Burns et al., 2002). Although discovered already a century ago, plaques and tangles are, till today, still the defining criteria for a definite post-mortem diagnosis.
It has been estimated that ∼5% of the population older than 65 years is affected by Alzheimer's disease (Bullock, 2004). The prevalence doubles approximately every 5 years beyond age 65 (Cummings, 2004) and some studies suggest that nearly half of the people aged 85 years and older suffer from this devastating disorder (Forsyth and Ritzline, 1998).
Due to the demographic development of Western societies, undoubtedly the number of patients and the economic impact of Alzheimer's disease will grow extraordinarily in the future without advances in therapy or prevention.
Current medications that have passed FDA approval for the treatment of Alzheimer's disease include acetylcholinesterase (AchE) inhibitors for mild to moderate cases, and memantine, an NMDA (N-methyl-D-aspatarte)-receptor antagonist for the treatment of moderate to severe Alzheimer dementia. All of these drugs seem to be able to produce modest symptomatic improvements in some of the patients (for review, seeClark and Karlawish, 2003; Cummings, 2004; Scarpini et al., 2003), none of the available medications, however, appears to be able to cure Alzheimer's dementia or to stop the disease progression.
There is enormous medical need for the development of novel therapeutic strategies that target the underlying pathogenic mechanisms in Alzheimer's disease and that are therefore expected to lead to new medications with strong disease-modifying properties.
Current status: symptomatic strategies
Cholinergic deficit
According to the ‘cholinergic hypothesis of Alzheimer's dementia’ the destruction of cholinergic neurons in the basal forebrain and the resulting deficit in central cholinergic transmission contribute substantially to the characteristic cognitive and non-cognitive symptoms observed in the patients (Bartus et al., 1982; Cummings and Back, 1998). Reductions in the activities of choline acetyltransferase and AChE in brain tissues from Alzheimer's disease patients were first reported in 1976 and 1977 (Bowen et al., 1976; Davies and Maloney, 1976; Perry et al., 1977). These enzymes are involved in the synthesis and degradation of acetylcholine, and the observed reduction in Alzheimer's disease suggested a selective destruction of cholinergic neurons. The cholinergic hypothesis provided the rational basis for the development of the AChE inhibitors for Alzheimer's disease therapy. Alternative approaches aiming for improved cholinergic neurotransmission, such as the administration of acetylcholine precursors, the stimulation of presynaptic acetylcholine release or muscarinergic agonists were not successful due to lack of efficacy or because of severe side effects (Doody et al., 2001). The acetylcholine deficiency hypothesis was primarily supported by post-mortem examinations of brains from patients with advanced dementia (Bartus et al., 1982; Perry, 1986; Whitehouse et al., 1986). The underlying assumption that the cholinergic deficits occur early in the course of the disease has been challenged by more recent studies reporting that the activities of the marker enzymes choline acetyltransferase and AChE were not reduced in individuals with mild Alzheimer's disease (Davis et al., 1999), and that cholinergic activity may be even up-regulated in early stage of the disease (DeKosky et al., 2002; Frolich, 2002).
Inhibition of brain cholinesterase activity
After its release into the synaptic cleft the neurotransmitter acetylcholine is degraded rapidly by the hydrolytic activity of cholinesterases. In the human brain, the most prominent enzyme involved in acetylcholine hydrolysis is AChE. Recent evidence suggests that additionally, butyrylcholinesterase (BChE) can also hydrolyse acetylcholine in the brain and may play a role in cholinergic transmission (Mesulam et al., 2002a, b).
Inhibition of these enzymes leads to an increase in the acetylcholine concentration in the synaptic cleft and is thus expected to enhance cholinergic transmission and ameliorate cholinergic deficit. Three different cholinesterase inhibitors, namely galantamine, donepezil and rivastigmine are commonly used for the treatment of mild to moderate Alzheimer's disease. Donepezil and galantamine are selective inhibitors of AChE, while rivastigmine also inhibits BChE, which accounts for ∼10% of the cholinesterase activity in normal human brain and appears to be predominantly associated with glia (reviewed in Scarpini et al., 2003).
Several randomized, double-blind, placebo-controlled studies reported positive effects of the cholinesterase inhibitors on cognitive and functional symptoms, as well as on behavioural abnormalities in Alzheimer's dementia (Rogers et al., 1998; Corey-Bloom, 1998; Rosler et al., 1999; Tariot et al., 2000; Winblad et al., 2001). Systematic reviews of the available randomized, double-blind, placebo-controlled studies by the Cochrane Collaboration support the use of the three cholinesterase inhibitors rivastigmine (Birks et al., 2000), donepezil (Birks and Harvey, 2003) and galantamine (Loy and Schneider, 2004) for treatment of mild to moderate Alzheimer's disease. The treatment effects observed at 6 months were moderate and of similar size for the three substances (reviewed in Scarpini et al., 2003). In line with the Cochrane reviews, clinical benefits from cholinesterase inhibitors were also reported in two other meta-analyses published in 2004 (Whitehead et al., 2004; Ritchie et al., 2004). In a recent systematic review, however, the scientific basis for the recommendations of cholinesterase inhibitors for treatment of Alzheimer's disease has been questioned (Kaduszkiewicz et al., 2005). Further long-term studies including the direct comparisons of the three cholinesterase inhibitors would be desirable.
Glutamate-mediated neurotoxicity
Glutamate excitotoxicity mediated through excessive activation of NMDA receptors is believed to play a role in the neuronal death observed in Alzheimer's disease and other neurodegenerative conditions (reviewed in Bleich et al., 2003; Hynd et al., 2004).
Glutamate represents the main excitatory neurotransmitter in the central nervous system and a physiological level of glutamate-receptor activity is essential for normal brain function (Kornhuber and Weller, 1997). Glutamate receptors can be broadly divided into metabotropic glutamate receptors, which are coupled to G-proteins, and ionotropic receptors, which are ligand gated ion channels. On the basis of their sensitivity to synthetic agonists, the latter are classified into the NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate (AMPA) and kainate receptors (Javitt, 2004).
In Alzheimer's disease, excessive activation of NMDA receptors is believed to cause increases in intracellular Ca2+ which then triggers downstream events that ultimately lead to neurodegeneration (for review, seeHynd et al., 2004). Consequently, NMDA-receptor antagonists may have a therapeutic potential for protecting neurons from glutamate-mediated neurotoxicity.
Potent NMDA-receptor antagonists like MK-801 or phencyclidine (PCP) were reported to produce psychotomimetic side effects (Kornhuber and Weller, 1997), presumably due to interference with the physiological functions of NMDA glutamate receptors. Memantine is a non-competitive NMDA-receptor antagonist with moderate affinity (Kornhuber et al., 1989) that appears to be able to protect neurons while leaving physiological NMDA-receptor activation unaffected (reviewed in Sonkusare et al., 2005). Memantine interacts with the NMDA receptor at therapeutic concentrations (Kornhuber and Quack, 1995).
Memantine was approved in 2002 in Europe for the treatment of ‘moderately severe to severe Alzheimer's disease’ and in 2003 in the United States for the treatment of moderate to severe cases of Alzheimer's disease (Sonkusare et al., 2005). A recent systematic review of double-blind, parallel group, placebo-controlled randomized trials of memantine in people with dementia published by the Cochrane Collaboration suggested a beneficial effect of memantine on cognitive function and functional decline in patients with moderate to severe Alzheimer's disease, and on cognitive function in vascular dementia. The drug was reported to be well-tolerated (Areosa Sastre et al., 2005).
Combination therapy
The positive clinical results of memantine monotherapy and the observation that memantine does not interact in vitro with the AChE inhibitors donepezil, galantamine or tetrahydroaminoacridine (Wenk et al., 2000) suggested that the clinical combination of memantine with cholinesterase inhibitors might represent a particularly valuable approach. A randomized, double-blind, placebo-controlled clinical trial of patients with moderate to severe Alzheimer's dementia who had already been adjusted to donepezil was published in January 2004. After 24 weeks, a statistically significant benefit of the combination therapy as compared with the monotherapy was observed with regard to measures of cognitive function, activities of daily living, behaviour and clinical global status (Tariot et al., 2004).
Mechanism-based therapeutic approaches targeting β-amyloid and tau pathologies
The characteristic neuropathological hallmarks of Alzheimer's disease include neuritic plaques and NFTs (Alzheimer, 1907, 1911). Neuritic plaques are extracellular lesions composed of a central core of aggregated amyloid-β peptide (Aβ) surrounded by dystrophic neurites, activated microglia and reactive astrocytes (Selkoe, 1991). In 1984, Glenner and Wong first reported on the purification and partial amino acid sequence determination of the β-amyloid peptide from cerebrovascular amyloid associated with Alzheimer's disease (Glenner and Wong, 1984). Shortly after, the 4 kDa amyloid protein components purified from the plaque cores from Alzheimer's disease and Down syndrome brains were found to be essentially identical, indicating a common origin (Masters et al., 1985)
NFTs are intracellular bundles of paired helical filaments (PHFs; Kidd, 1963; Terry, 1963) and straight filaments (Yagishita et al., 1981). They are composed of tau protein (Delacourte and Defossez, 1986; Grundke-Iqbal et al., 1986a; Kosik et al., 1986; Goedert et al., 1988; Wischik et al., 1988) in an abnormally hyperphosphorylated form (Grundke-Iqbal et al., 1986b; Flament et al., 1989; Lee et al., 1991; Goedert et al., 1992; Hasegawa et al., 1992; Sergeant et al., 1995). It appears that these two proteinacious lesions are at the root of the pathogenesis of Alzheimer's disease, and consequently it is believed that targeting the underlying mechanisms leading to plaques and tangles will ultimately generate novel therapeutics with disease-modifying properties.
Therapeutic strategies targeting β-amyloid
The amyloid cascade hypothesis
The dominating hypothesis to explain the mechanisms leading to Alzheimer's disease is the amyloid cascade hypothesis, which states that the Aβ, a fragment of the amyloid precursor protein (APP), plays a central role in the pathogenesis. Aβ is produced proteolytically from APP by the so called β- and γ-secretases. It is believed that accumulation of β-amyloid (in particular of the Aβ42 peptide) in the brain initiates a cascade of events that ultimately leads to neuronal dysfunction, neurodegeneration and dementia (Fig. 1; for a review, seeHardy and Selkoe, 2002).
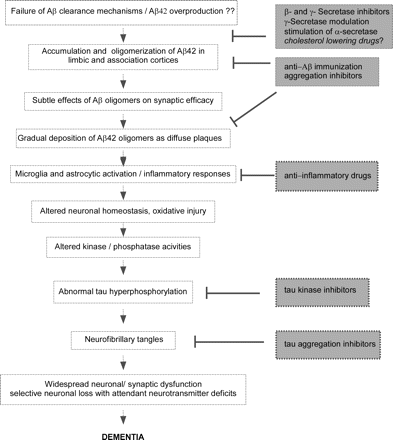
Amyloid cascade hypothesis and selected strategies for therapeutic intervention. The figure summarizes the presumed sequence of pathological processes that leads to neurodegeneration in AD according to the amyloid cascade hypothesis (Hardy and Selkoe, 2002), and indicates selected potential approaches for therapeutic intervention. Aβ42 is believed to initiate this series of pathogenic events. Modified from: D. Selkoe, ‘The amyloid hypothesis’. Alzheimer Research Forum. Available at Author Webpage. Accessed in August 2006.
The strongest argument supporting a causal role of β-amyloid in Alzheimer's disease comes from the identification of mutations in the APP gene (Chartier-Harlin et al., 1991; Goate et al., 1991; Murrell et al., 1991) and in the genes for presenilin-1 and -2 (PS1 and PS2; Levy-Lahad et al., 1995; Sherrington et al., 1995) that are responsible for early-onset forms of familial Alzheimer's disease (FAD). By July 2006, 25 pathogenic mutations in APP, 155 in PS1 and 10 in PS2 were listed on the Alzheimer Disease & Frontotemporal Dementia Mutation Database (Cruts and Rademakers, 2006; Author Webpage). Another online database listing FAD mutations is available at Author Webpage. FAD mutations in PS1 and PS2, as well as mutations in the APP gene close to the γ-secretase cleavage site, modify the proteolytic generation of Aβ peptides in such a way that the relative proportion of the highly amyloidogenic Aβ42 form is increased (Suzuki et al., 1994; Tamaoka et al., 1994; Borchelt et al., 1996; Duff et al., 1996; Citron et al., 1997). The so-called ‘Swedish’ APP double mutation (KM670/671NL) leads to a rise in overall Aβ generation due to an increased cleavage by β-secretase (Citron et al., 1992; for reviews, seeHardy and Selkoe, 2002; St George Hyslop and Petit, 2004). Aβ peptides represent the principal protein component of the neuritic plaques characteristic for Alzheimer's disease and it was shown that aggregated forms of synthetic Aβ peptides can cause damage to cultured neuronal cells (Pike et al., 1993; Lorenzo and Yankner, 1994). More recent findings suggest that rather than highly aggregated Aβ species, soluble oligomeric prefibrillar forms of Aβ [so called Aβ-derived diffusible ligands (ADDLs) or protofibrils] may represent the neurotoxic entity and cause synaptic dysfunction (Lambert et al., 1998; Hartley et al., 1999).
Transgenic animal models may help to better understand the role of amyloid and tau in the aetiology of Alzheimer's disease and they may also serve for testing novel drug candidate compounds.
Transgenic mice that show robust amyloid plaque pathology were first reported by Games and colleagues in 1995 (Games et al., 1995). These mice expressed high levels of the V717F FAD-mutant form of human APP and developed extracellular amyloid plaques, astrocytosis and neuritic dystrophy. In 1996, transgenic ‘Tg2576’ mice over-expressing the Swedish APP double mutation were shown to develop Congo red positive amyloid plaques and age-dependent correlative memory deficits (Hsiao et al., 1996). Sturchler-Pierrat and colleagues (1997) generated the APP23 line expressing Swedish mutant APP under control of the Thy-1 promoter. These mice developed typical plaques and showed signs of inflammatory reactions as well as cerebrovascular amyloid deposits (Sturchler-Pierrat et al., 1997; Calhoun et al., 1999). At age 14–18 months, a selective reduction of neurons in the hippocampal area CA1 was observed (Calhoun et al., 1998). (For a recent review on additional mouse lines that have been generated since then, seeMcGowan et al., 2006.)
According to the amyloid cascade hypothesis novel therapeutic strategies that lower Aβ levels or prevent the formation of the presumed neurotoxic oligomeric Aβ species are predicted to stop or slow down the progession of neurodegeneration and dementia in Alzheimer's disease.
Modulation of Aβ production
Aβ peptides are proteolytic fragments of the APP, a large integral membrane protein that is composed of a signal sequence, a large extra-membranous region, a single transmembrane domain and a small cytosolic C-terminal tail (Kang et al., 1987). Post-translational modifications of APP include phosphorylation, tyrosine-sulphation and N- and O-linked glycosylations (Oltersdorf et al., 1990; Weidemann et al., 1989). Aβ is generated from APP by sequential cleavages by two proteases termed β- and γ-secretase. APP cleavage by the so-called α-secretase, which was the first proteolytic cleavage to be identified, precludes Aβ generation since the α-secretase cleavage site is located within the Aβ sequence (Esch et al., 1990; Sisodia et al., 1990). Aβ is not the result of abnormal or pathological APP processing, as was originally believed, but is secreted constitutively by normal cells in culture (Haass et al., 1992; Shoji et al., 1992) and can be detected in plasma and CSF of healthy humans (Seubert et al., 1992). The observation that γ-secretase activity was prevented in neuronal cells derived from PS1 deficient mouse embryos indicated that PS was tightly linked to the intramembrane cleavage of APP (De Strooper et al., 1998). Two conserved aspartate residues in PS1 located in transmembrane regions were shown to be essential for γ-secretase activity (Wolfe et al., 1999), and subsequent studies revealed that γ-secretase is a protein complex composed of PS, nicastrin, PEN2 and APH-1. It appears that PS1 provides the active core of the secretase complex and that the enzymatic mechanism is that of an aspartate protease (reviewed in De Strooper, 2003).
Beta-secretase was discovered and cloned in 1999 (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999) and has been a major focus of drug discovery efforts since then. BACE1 knock-out mice were reported to produce only very small amounts of Aβ confirming that BACE1 represents the primary β-secretase in vivo. Furthermore, the absence of severe phenotypes in the knockout mice (Luo et al., 2001; Roberds et al., 2001), suggests that targeting β-secretase may be a particularly promising therapeutic approach, even though the identification of specific small molecule inhibitors suitable for drug development appears to be difficult (Citron, 2004).
Several pharmaceutical companies have actively searched for small molecule compounds that can reduce Aβ production by affecting one of these targets.
A γ-secretase inhibiting compound (LY450139) by Eli Lilly was recently tested in a 6-week Phase II trial. The compound was reported to reduce Aβ levels in plasma but not in CSF at concentrations that did not produce significant side effects (Siemers et al., 2005).
A major concern regarding the therapeutic usefulness of γ-secretase inhibition and potential side effects comes from the identification of several γ-secretase substrates other than APP, including Notch 1 and others (for review, seeDe Strooper, 2003).
The finding that certain non-steroidal anti-inflammatory drugs (NSAIDs) can preferentially reduce the generation of the highly amyloidogenic Aβ42 species without affecting Notch cleavage (Weggen et al., 2001), indicates the existence of a γ-secretase modulating mechanism as a potential drug target that may allow for lowering Aβ42 levels without inducing potential side effects related to complete inhibition of γ-secretase. It is reasonable to assume that currently more potent and specific Aβ42 lowering compounds are being actively searched for.
Cleavage of APP by non-amyloidogenic α-secretase can be stimulated by muscarinic acetylcholine-receptor agonists, and this was shown to also reduce Aβ generation in cell culture (Hung et al., 1993; Wolf et al., 1995). M1 muscarinic acetylcholine-receptor agonists were therefore suggested to be potentially useful not only for symptomatic treatment of Alzheimer's disease but to a limited extent also for causal therapy (Fisher, 2000).
The M1 agonist AF267B (Fisher, 2000) was recently tested in triple-transgenic mice expressing mutant forms of presenilin 1, APP and tau (Oddo et al., 2003; Billings et al., 2005). A 10-week treatment of the mice, with daily intraperitoneal injections of the compound, was reported to ameliorate cognitive deficit in the mice and to reduce both, amyloid and tau pathologies (Caccamo et al., 2006).
Inhibition of Aβ-aggregation
Preventing the formation of the presumed toxic oligomeric aggregates of Aβ by small molecules represents another promising approach for the development of novel and causal therapeutics for treating Alzheimer's disease.
Neurochem Inc., a Canadian company, has completed a Phase II clinical trial of their glycosaminoglycan mimetic Alzhemed that has been designed to bind to Aβ peptides and thereby inhibits formation of Aβ aggregates. A phase III trial is planned (reviewed in Citron, 2004).
Metal ions like Cu2+ and Zn2+ may be involved in the mediation of Aβ aggregation and toxicity (Atwood et al., 1998). A significant decrease in brain Aβ deposition in APP-transgenic mice was observed after 9 weeks treatment with clioquinol, an antibiotic and Cu/Zn chelator that crosses the blood–brain barrier (Cherny et al., 2001). Recently Prana Biotechnology cancelled an upcoming Phase II/III clinical trial of clioquinol (PBT-1) because of toxic impurities believed to occur during the manufacture (Boggs, 2005; Prana Biotechnology, 2005).
Aβ immunotherapy
In a landmark paper in 1999 Dale Schenk and co-workers described that immunization with Aβ attenuates the Alzheimer's disease-like pathology in a transgenic mouse model of Alzheimer's disease (Schenk et al., 1999). Using peripheral antibody administration the same group provided direct evidence that Aβ antibodies are sufficient to reduce the amyloid deposition (Bard et al., 2000). These fundamental observations have meanwhile been confirmed in different transgenic Alzheimer's disease models as well as in aged non-human primates, which develop some brain amyloid in particular cerebral amyloid angiopathy (CAA; Lemere et al., 2004). Furthermore, Aβ immunization was shown to also reduce various aspects of the amyloid-associated pathology including neuritic dystrophy and synaptic degeneration as well as early tau accumulation (Lombardo et al., 2003; Oddo et al., 2004; Brendza et al., 2005; Buttini et al., 2005).
These histopathological normalizations also result in functional improvements. Active and passive immunization against Aβ can reduce the learning deficits of APP-transgenic mice (Janus et al., 2000; Morgan et al., 2000). An amelioration of memory deficits can already be found after short term and even a single passive immunization in the absence of an amyloid reduction (Dodart et al., 2002; Kotilinek et al., 2002). This lack of correlation with amyloid deposits probably reflects the fact that some behavioural deficits seem to be induced by amyloid deposits while others may be more acutely caused by soluble Aβ species (oligomers). In accordance Aβ immunization has been demonstrated to neutralize infused Aβ oligomers and to improve synaptic plasticity impaired by these oligomers (Hartman et al., 2005; Klyubin et al., 2005).
Three different, though not mutually exclusive, mechanisms have been proposed to explain the amyloid lowering effect of Aβ immunization. Following the detection of antibodies bound to brain amyloid deposits it has been postulated that they trigger Fc-receptor-mediated phagocytosis (Schenk et al., 1999; Bard et al., 2000). Compatibly, microglia activation, increased Fcγ-receptor expression and a superior efficacy of IgG2a antibodies showing highest Fcγ-receptor affinity have been observed (Schenk et al., 1999; Bacskai et al., 2001; Bard et al., 2003; Wilcock et al., 2003; Bussiere et al., 2004; Wilcock et al., 2004b). In addition, in vivo efficacy of Aβ antibodies correlated with their ability to induce phagocytosis in an in vitro system (Bard et al., 2000). As an alternative mechanism, the antibodies might act as chaperones and disrupt Aβ aggregates or prevent aggregation (Solomon et al., 1997). Supporting this hypothesis, antibodies can block and even reverse Aβ aggregation and toxicity in vitro (Solomon et al., 1997; Frenkel et al., 2000; McLaurin et al., 2002; Du et al., 2003). In vivo Fc-receptor independent clearance of amyloid deposits has been observed with F(ab′)2 fragments and in a Fcγ-receptor knock-out background (Bacskai et al., 2002; Das et al., 2003; Wilcock et al., 2004a). While evidence for both hypotheses seems contradictory, a possible explanation comes from a study describing a rapid microglia-independent clearance of diffuse amyloid followed by a microglia-dependent elimination of compact plaques (Wilcock et al., 2003). Finally, circulating antibodies were postulated to sequester Aβ, shift the equilibrium towards the periphery and thereby reduce brain Aβ deposition (DeMattos et al., 2001). Consistent with this peripheral sink hypothesis an elevation of blood Aβ after immunization has been found (DeMattos et al., 2001; Pfeifer et al., 2002; Lemere et al., 2003; Gandy et al., 2004; Lemere et al., 2004; Wilcock et al., 2004b) which reflected the brain amyloid burden (DeMattos et al., 2002). Yet, this could also be explained by a simple stabilization of blood Aβ due to antibody binding. At present it is not possible to exclude any of the three hypothetical action mechanisms as they may act in concert and depend on the particular experimental paradigm (e.g. level of Aβ generation, isoform ratios and amyloid type, as well as, stage of amyloid formation or route of administration). More studies, which better consider these parameters, will be needed to determine their relative contribution to the overall effects.
The first clinical trials of Aβ immunotherapy, which used aggregated Aβ1–42 as antigen, had to be stopped in Phase II due to aseptic meningoencephalitis in 6% of the treated patients (Orgogozo et al., 2003; Bayer et al., 2005; Gilman et al., 2005). Autopsy studies of two affected patients demonstrated a T-cell-mediated autoimmune response (Ferrer et al., 2004; Nicoll et al., 2003) presumably directed against Aβ. The use of full-length Aβ containing T-cell epitopes (Monsonego et al., 2003) with a strong T-cell adjuvant (QS21; Cribbs et al., 2003) and the supplementation of the vaccine by polysorbate-80 (Tween-80) during the Phase II trial (Gilman et al., 2005) may have contributed to the adverse response. Evidence for efficacy of Aβ immunotherapy was obtained in the first three autopsies, which showed extensive neo-cortical areas devoid of amyloid plaques and associated dystrophic neurites and astrocytes, while amyloid angiopathy and the NFTs were not reduced (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). Clinically, antibody responders significantly improved over 1 year in some memory tests, while others did not change significantly (Gilman et al., 2005). In a small subset tested for CSF tau a significant decrease was found indicative of a reduced degeneration. MRI detected greater brain volume decreases and ventricular enlargements in antibody responders, which is not understood but the amyloid removal may directly or indirectly be responsible for this effect (Fox et al., 2005). Considering the limitations of the study, as well as the positive trends in several efficacy measures, additional testing of Aβ immunotherapy seems warranted if the safety issues can be addressed.
Extensive studies of active Aβ immunization in mice and other species had not predicted autoimmune disease although meningoencephalitis (Lee et al., 2005a) as well as an elevation in cerebral haemorrhages (Pfeifer et al., 2002; Wilcock et al., 2004c; Racke et al., 2005) has meanwhile been described after passive immunization. While the significance of these findings with respect to the adverse events in the active immunization study in humans remains open, the findings need to be considered in the development of alternative approaches. These mainly aim to avoid the unwanted T-cell response. For active immunization alternative adjuvants (Cribbs et al., 2003; Maier et al., 2005), use of the mucosal immune system (Weiner et al., 2000; Leverone et al., 2003) or of Aβ fragments (Li et al., 2004c; Agadjanyan et al., 2005; Solomon, 2005; Zurbriggen et al., 2005) are exploited. The Aβ peptides used span the B-cell epitopes in the N-terminal part and are linked to carrier proteins including viral structures or other independent T-cell epitopes, which should not induce an Aβ-specific T-cell response. Passive Aβ immunotherapy with monoclonal antibodies is being evaluated, as well as DNA vaccines expressing Aβ and fragments thereof. If these second generation approaches show the expected safety profile Aβ immunotherapy holds promise as a disease-modifying Alzheimer's disease therapy.
Therapeutic strategies targeting tau hyperphosphorylation and neurofibrillary degeneration
Neurofibrillary lesions made up from aggregated hyperphosphorylated forms of the microtubule-associated protein tau represent a second defining neuropathological feature of Alzheimer's disease. The pathological hyperphosphorylation of tau, which can be visualized by immunochemical methods, is an early event in the development of Alzheimer's disease-related neurofibrillary changes (Braak and Braak, 1995). Phosphorylation of tau regulates its ability to promote microtubule assembly (Lindwall and Cole, 1984) and abnormal hyperphosphorylation interferes with its normal biological function (Gustke et al., 1992; Bramblett et al., 1993; Alonso et al., 1994) by decreasing tau's ability to bind to, and to stabilize, microtubules. This loss of function can be restored in vitro by dephosphorylation of pathological tau protein with phosphatases (Iqbal et al., 1994). Under pathological conditions, an imbalance of kinase and phosphatase activities may lead to aberrant hyperphosphorylation of tau resulting in its detachment from microtubules, breakdown of the microtubule network, disturbance of axonal transport and ultimately neurodegeneration (Mandelkow and Mandelkow, 1998; Fig. 2). Additionally, certain pathological forms of tau may also have direct neurotoxic properties (‘gain of toxic function’; Shahani and Brandt, 2002). The identification of mutations in the tau gene that are responsible for familial frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) indicated that malfunction or dysregulation of tau alone can be sufficient to induce neurodegeneration (Hutton et al., 1998; Spillantini et al., 1998). Until now, 40 different pathogenic tau mutations have been reported that cause frontotemporal dementia (Cruts and Rademakers, 2006; Alzheimer Disease and Frontotemporal Dementia Mutation Database; available at Author Webpage). The neuropathology in these cases is characterized by neuronal loss and the presence of neuronal or neuronal and glial aggregates of hyperphosphorylated tau protein (Lee et al., 2001; Dermaut et al., 2005). The molecular details of tau-related neurodegeneration and the identity of the presumed neurotoxic species are not well understood, yet. Recent findings in transgenic mice expressing non-mutant human tau isoforms, suggest that neuronal death may not be directly linked to the formation of the highly aggregated NFTs (Andorfer et al., 2005). In line with these observations, Santacruz and co-workers reported functional improvements but ongoing NFT formation in transgenic mice after suppression of mutant human tau expression (Santacruz et al., 2005).
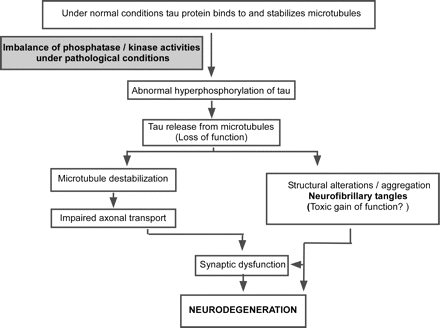
A hypothetical sequence of events leading to neurofibrillary degeneration in Alzheimer's disease: under pathological conditions (possibly triggered by oligomeric Aβ) an imbalance of phosphatase and kinase activities results in abnormal hyperphosphorylation of tau protein. Release of hyperphosphorylated tau protein destabilizes microtubules which affects axonal transport and leads to synaptic dysfunction and degeneration. Unbound tau protein can aggregate and form NFTs. Hyperphosphorylated and/or aggregated tau species may have direct neurotoxic effects (‘toxic gain of function’).
The inhibition of tau-related neurofibrillary degeneration represents a highly promising approach in search for novel therapies for Alzheimer's disease and related tauopathies. This may be achieved by targeting one or more tau kinase(s), by increasing the activity of protein phosphatase (PP)-2A or by inhibition of the presumed toxic properties of pathological tau proteins.
Inhibition of tau kinases
More than 30 phosphorylation sites on tau protein have been described and numerous proline directed and non-proline directed kinases were shown to be able to phosphorylate tau protein in vitro. These include glycogen synthase kinase 3-β (GSK3-β), cdc2-like kinase (cdk5), extracellular signal-regulating kinase-2 (ERK2), microtubule-affinity-regulating kinase (MARK), protein kinase A (PKA), members of the stress-activated protein kinase (SAPK) family, Ca2+/calmodulin-dependent kinase II and casein kinases I and II (for reviews seeJohnson and Hartigan, 1998; Buee et al., 2000).
While it is clear that aberrant phosphorylation of tau protein is a key feature of neurofibrillary degeneration, the exact role of particular phosphorylation sites on tau and the identity of the relevant protein kinases that contribute to their phosphorylation under pathological conditions remain elusive.
Of the many potential tau kinases, GSK3β and cdk5/p25 have received particular attention. Cruz et al. (2003), reported that inducible over-expression of the cdk5 activator p25 in the postnatal forebrain of transgenic mice resulted in tau hyperphosphorylation and aggregation as well as in neuronal loss, providing strong evidence that aberrant kinase activity can lead to neurodegeneration. When transgenic mice over-expressing p25 were crossed with mice transgenic for human tau carrying the P301L FTDP-17 mutation, an increase in tau hyperphosphorylation and aggregation relative to P301L tau single transgenic mice was observed. Interestingly, in these double transgenic mice insoluble tau was associated with activated GSK3, suggesting that although p25/cdk5 provided the initial trigger, at least one additional kinase (GSK3) appeared to be involved (Noble et al., 2003). While sarcosyl-insoluble hyperphosphorylated tau was increased in these double transgenic mice, this was apparently not associated with significantly accelerated dystonia as compared with the P301L tau single transgenic mice.
Neuronal inducible over-expression of GSK3-β in hippocampus and cortex of transgenic mice was shown to increase tau phosphorylation at the PHF1 epitope, to induce somatodendritic localization of tau and to lead to neurodegeneration (Lucas et al., 2001). While these observations clearly support GSK3-β as a tau kinase in vivo, its role in the tau-related pathology remains somehow controversial: in double transgenic mice expressing wild-type human tau and a constitutively active form of GSK3-β, a 2-fold increase in GSK3-β kinase activity appeared to reduce the neuropathology and motor impairments that were observed in single tau transgenic mice, (Spittaels et al., 2000).
Another candidate kinase that has been implicated in abnormal hyperphosphorylation of tau is the MAP kinase ERK2, which can phosphorylate tau in vitro at many of the Ser/Thr-Pro motifs and to high stoichiometry (Roder and Ingram, 1991; Drewes et al., 1992). Importantly, ERK2 and several members of the SAPK family but not GSK3 and cdk5 (neuronal cdc2-like kinase) were shown to be able to phosphorylate tau at Ser422, which is one of very few phosphorylation sites that appear to be specific for disease (Hasegawa et al., 1996; Goedert et al., 1997). Activated forms of ERK1/2 and the upstream activating kinases MEK1/2 were shown to co-distribute with the progressive neurofibrillary changes in Alzheimer's disease (Pei et al., 2002; Perry et al., 1999).
In cell-culture experiments, however, stimulation of MAP kinase by v-raf transformation did not induce tau hyperphosphorylation (Latimer et al., 1995), nor did inhibitors of the classical MEK–ERK activation pathway prevent tau hyperphosphorylation in cellular models involving okadaic acid (Ho et al., 1997) or arsenite (Giasson et al., 2002).
Several animal models have been developed, that reproduce characteristic features of tau-related neurofibrillary degeneration and that may serve for testing novel kinase inhibitors in vivo to evaluate their therapeutic potential and to assess the role of particular kinases in tau filament formation and neurodegeneration. These models include for example transgenic mice expressing FTDP-17 mutant forms of human tau (Lewis et al., 2000; Gotz et al., 2001) as well as novel triple-transgenic mice developing both, tau and amyloid pathologies (Oddo et al., 2003).
Recently, Noble and co-workers reported that chronic inhibition of GSK3 for 30 days in vivo by lithium reduced tau hyperphosphorylation at several sites and decreased the levels of aggregated insoluble tau in JNPL3 transgenic mice over-expressing mutant human tau (Noble et al., 2005). Strong in vivo evidence that inhibition of pathological tau hyperphosphorylation can also have a functional impact and therefore represents a particularly promising therapeutic strategy comes from a very recent study. Le Corre and co-workers treated JNPL3 mice transgenic for P301L mutant tau for 9 weeks with a novel orally available and blood–brain-barrier-penetrating synthetic kinase inhibitor. The compound was selected from a series of synthetic indolocarbazoles with limited kinase selectivity but capable of preventing tau hyperphosphorylation in cell and brain slice culture models. A significant delay in the onset of the typical motor deficits in these mice was observed in the treated group as compared to the controls, and this was accompanied by a reduction in abnormal tau hyperphosphorylation (Le Corre et al., 2006). Taken together, these observations strongly support the use of inhibitors of aberrant phosphorylation of tau as an approach to developing a disease-modifying treatment for Alzheimer's disease and other tau-related neurodegenerative diseases.
Prolyl-isomerase Pin1
In 1999, Lu et al. discovered that the peptidyl prolyl cis/trans isomerase Pin1 bound to tau protein phosphorylated at Thr231 and co-purified with PHFs from Alzheimer's disease brain. In vitro, Pin1 was shown to restore the ability of phosphorylated tau to promote microtubule assembly (Lu et al., 1999). Additionally, Pin1 can facilitate dephosphorylation of tau by phosphatase PP2A (Zhou et al., 2000). Pin1 knockout mice were reported to develop tau hyperphosphorylation, sarcosyl-insoluble filamentous tau aggregates and neuronal degeneration in an age-related fashion (Liou et al., 2003). These observations suggest that Pin 1 may have protective functions against age-related neurodegeneration (Lu, 2004). Ramakrishnan et al. (2003) reported the detection of Pin1 granules in early stages of Alzheimer's disease, FTDP-17 (P301L) and Pick's disease and discussed several different possible scenarios concerning Pin1's role in tauopathies. One of these suggested that Pin1 may be involved in the pathogenesis and may promote the development of neurofibrillary pathology. Understanding the exact role of Pin1 in disease will be a prerequisite to evaluate Pin1 as a potential novel therapeutic target.
Activation of phosphatases
The phosphorylation state of any phosphoprotein results from the activities of both, kinases and phosphatases. It has been suggested, that in Alzheimer's disease, an imbalance of kinase and phosphatase activities may lead to abnormal hyperphosphorylation of tau protein (Mandelkow and Mandelkow, 1998). Reduced activites of tau-phosphatases have been reported in Alzheimer's disease brain as compared to controls (Gong et al., 1995). Protein phosphatases PP2A, PP2B and, to a lesser extent PP1, can dephosphorylate tau protein in vitro (reviewed in Lau et al., 2002). Additionally, PP2A was also shown to be involved in the regulation of tau phosphorylation in vivo (Gong et al., 2000). Expression of a dominant negative form of PP2A in transgenic mice under control of a neuron-specific promoter resulted in a 34% reduced activity of PP2A, and induced tau hyperphosphorylation at Ser202/Thr205 and Ser422 (Kins et al., 2001).
Thus, it has been suggested that in addition to kinase inhibition, restoration or up-regulation of tau phosphatase activities (e.g. PP2A) may represent another potential approach to inhibition of abnormal tau hyperphosphorylation (Iqbal and Grundke-Iqbal, 2004).
Memantine, an NMDA-receptor antagonist approved for the treatment of moderate to severe Alzheimer's disease was recently reported to inhibit okadaic acid-induced abnormal tau hyperphosphorylation and the associated neurodegeneration in rat hippocampal slices. Interestingly, it was suggested that memantine exerted this effect by restoration of PP2A activity through ‘PP2A signalling’ (Li et al., 2004b).
Inhibition of tau aggregation
Filamentous tau lesions in the affected brain regions represent the defining neuropathological features of tauopathies (for reviews, seeTolnay and Probst, 1999; Lee et al., 2001). In Alzheimer's disease, the intraneuronal NFTs contain PHFs as the major and straight filaments as a minor component, both of which are composed of hyperphosphorylated tau proteins (see above). The neurofibrillary lesions in Alzheimer's disease develop in a predictable spatiotemporal sequence, and the six stages of disease progression have been defined by Braak and Braak (1991, 1995). NFTs were shown to correlate with neuronal loss (Fukutani et al., 1995; Gomez-Isla et al., 1997) and with severity of dementia (Arriagada et al., 1992; Wilcock and Esiri, 1982). The hypothesis that tau aggregation and NFT formation are directly linked to neurodegeneration is supported by recent observations from cultured neuroblastoma cells inducibly over-expressing tau fragments. Only those mutant tau fragments that formed aggregates but not soluble forms were found to be cytotoxic (Khlistunova et al., 2006). Thus, substances that can inhibit tau aggregation might have the potential to ultimately protect neurons from neurofibrillary degeneration. Methods for screening for tau aggregation inhibitors have been developed and potential small molecule candidate compounds have been identified (Chirita et al., 2004; Pickhardt et al., 2005).
At present, however, the exact properties of the presumed neurotoxic form of abnormal tau protein and the precise role of hyperphosphorylation and aggregation in the pathological processes are not clear. Recent findings in mice transgenic for wild-type or mutant human tau indicate that tau-related neurodegeneration can occur independently of NFT formation and that NFTs do not invariably cause neuronal loss (Andorfer et al., 2005; Santacruz et al., 2005). It has also been proposed that aggregation of hyperphosphorylated tau into PHFs may represent a protective mechanism to sequester toxic forms of abnormal tau protein (Lee et al., 2005b). A similar protective function of protein aggregation has been shown for huntingtin (Arrasate et al., 2004).
Other approaches
Markers of neuroinflammation including activated microglia and astrocytes, complement components and inflammatory cytokines are typically observed in association with Alzheimer's disease neuropathology (for review, seeMcGeer and McGeer, 2003; Tuppo and Arias, 2005). Observational retrospective and prospective studies indicated that the long-term use of NSAIDs may have a preventive effect against the development of Alzheimer's disease (reviewed in Szekely et al., 2004) suggesting that neuroinflammation may contribute to the neurodegeneration.
The selective cyclooxygenase (COX)-2 inhibitor rofecoxib and the non-selective NSAID, naproxen, were also tested in a clinical randomized control trial for the treatment of mild to moderate Alzheimer's disease, but neither drug was able to slow the rate of cognitive decline as compared with the placebo control group (Aisen et al., 2003). Some NSAIDs including ibuprofen can modify γ-secretase activity in such a way that, specifically, the production of Aβ42 peptides is decreased (see above and Weggen et al., 2001). In APP-transgenic mice, ibuprofen reduced amyloid load and microglial activation (Lim et al., 2000) suggesting an effect at an early stage of plaque pathology.
Cholesterol metabolism appears to play an important role in the biology of APP and possibly also in the pathological processes leading to Alzheimer's disease. APP processing and Aβ production are sensitive to cholesterol levels (Simons et al., 1998). The activities of both, β- and γ-secretase, were shown to be inhibited by lowering cholesterol in cultured neurons (Cordy et al., 2003; Wahrle et al., 2002). Treatment with cholesterol lowering drugs reduced Aβ levels in vivo in cerebrospinal fluid of guinea pigs (Fassbender et al., 2001) and alleviated Aβ pathology in transgenic mice (Refolo et al., 2001). In humans, lovastatin was reported to reduce serum Aβ concentration in a dose-dependent manner (Friedhoff et al., 2001; cited in Wolozin, 2004). Retrospective epidemiological studies indicated a reduced risk of developing dementia in patients taking statins (Jick et al., 2000; Wolozin et al., 2000). In contrast, three prospective studies failed to show a protective effect of statins with regard to cognitive function (Shepherd et al., 2002; Heart Protection Study Collaborative Group, 2002; Li et al., 2004a). Interestingly, elevated plasma cholesterol levels were reported in individuals carrying the apolipoprotein epsilon 4 allele (APOE4; Sing and Davignon, 1985; Ehnholm et al., 1986), which is the major genetic risk factor for Alzheimer's disease (Corder et al., 1993; Poirier et al., 1993). At present, the exact mechanism by which APOE4 affects the pathophysiology of Alzheimer's disease is not clear. A recent meta-analysis did not reveal Alzheimer's disease associated polymorphisms in cholesterol-related genes other than APOE and it was therefore concluded that the link between Alzheimer's disease and APOE4 was probably not directly related to cholesterol (Wolozin et al., 2006).
Summary and conclusions
Medications for the treatment of Alzheimer's disease that are available today include cholinesterase inhibitors and the NMDA-receptor antagonist, memantine. These drugs are safe and in several large and independent studies, they were reported to produce moderate symptomatic benefits. At present, however, there is no treatment available that can stop the progressive deterioration of cognitive functions in the Alzheimer's disease patients. The development of novel drugs with strong disease-modifying properties therefore represents one of the biggest unmet medical needs today.
The pathophysiology of Alzheimer's disease and the search for novel therapeutic strategies have been a major focus of academic and industry research for several years. The predominant hypothesis to explain the pathogenesis is the amyloid cascade hypothesis, and consequently, several of the novel and promising therapeutic strategies are specifically addressing the amyloid pathology.
Whether anti Aβ-immunotherapy, small molecule secretase inhibitors, other Aβ lowering approaches or aggregation inhibitors will turn out to be safe and will be able to stop or slow down disease progression remains to be seen.
Conflict of interest
The authors would like to mention that M.S. is an employee of Novartis in Basle, Switzerland and H.-W.K. was an employee of NADAG and Sirenade Pharmaceuticals (the latter resulted from the merger of NADAG and Sireen AG) and was involved in research activities aiming for the discovery of kinase inhibitors as potential medications for Alzheimer's disease.
H.-W.K. is supported by the grant HBPP-NGFN2 (01 GR 0447) funded by the German Federal Ministry of Education and Science (BMBF). J.W. is supported by the BMBF funded grants Competence Net Dementias (01 GI 0420) and HBPP-NGFN2 (01 GR 0447). Funding to pay the Open Access publication charges for this article was provided by the BMBF funded grant Competence Net Dementias (01 GI 0420).


{kind=link}
{kind=link}