




Abstract
Male factor infertility concerns 7–10% of men and among these 40–60% remain unexplained.
This review is based on recent published literature regarding the genetic causes of male infertility.
Screening for karyotype abnormalities, biallelic pathogenic variants in the CFTR gene and Y-chromosomal microdeletions have been routine in andrology practice for >20 years, explaining ~10% of infertility cases. Rare specific conditions, such as congenital hypogonadotropic hypogonadism, disorders of sex development and defects of sperm morphology and motility, are caused by pathogenic variants in recurrently affected genes, which facilitate high diagnostic yield (40–60%) of targeted gene panel-based testing.
Progress in mapping monogenic causes of quantitative spermatogenic failure, the major form of male infertility, has been slower. No ‘recurrently’ mutated key gene has been identified and worldwide, a few hundred patients in total have been assigned a possible monogenic cause.
Given the high genetic heterogeneity, an optimal approach to screen for heterogenous genetic causes of spermatogenic failure is sequencing exomes or in perspective, genomes. Clinical guidelines developed by multidisciplinary experts are needed for smooth integration of expanded molecular diagnostics in the routine management of infertile men.
Di−/oligogenic causes, structural and common variants implicated in multifactorial inheritance may explain the ‘hidden’ genetic factors. It is also critical to understand how the recently identified diverse genetic factors of infertility link to general male health concerns across lifespan and how the clinical assessment could benefit from this knowledge.
Introduction
Infertility is defined as a failure to achieve a wanted pregnancy within a 12-month period despite regular unprotected sexual intercourse. Male factor infertility concerns 7–10% of men, representing a prevalent health condition with broad clinical and social consequences.1–3 Male infertility is not a single condition with uniform aetiology and treatment options, but rather a clinical endpoint of diverse pathological processes and sub-phenotypes (Fig. 1a). The palette of possible congenital conditions includes urogenital and gonadal anomalies, disturbances of hypothalamic–pituitary-gonadal axis, primary testicular failure causing quantitative impairment of spermatogenesis, ductal obstruction and qualitative sperm defects, such as abnormal sperm morphology and/or motility.3–5 Acquired factors leading to male infertility are, for example genitourinary infections and inflammations, oncological and chronic diseases, testicular damage due to traumas, operations, abuse of anabolic steroids, sexual dysfunction, etc.
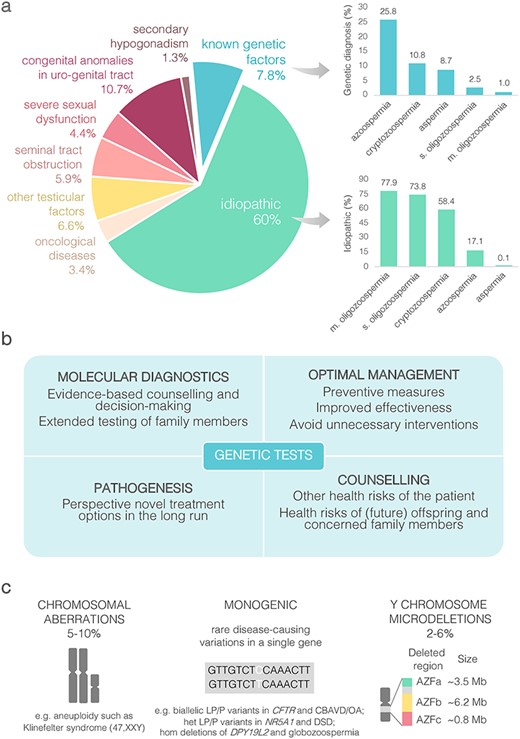
Status quo in genetics of male infertility. (a) Aetiology of male infertility and proportion of genetically diagnosed and idiopathic cases in the subgroups of patients with reduced sperm counts (based on data from4). (b) Added value of genetic testing. (c) Established genetic causes of male factor infertility. CBAVD, congenital absence of vas deferens; het, heterozygous; hom, homozygous; (L)P, (likely) pathogenic; m, moderate; s, severe.
Today, 40–60% of cases with spermatogenic impairment remain unexplained and among moderate oligozoospermia cases (sperm counts 10–39 million per ejaculate), this fraction is close to 80%. The high number of idiopathic cases refers to an urgent clinical need to improve the knowledge about the causes of infertility and translate the findings into routine diagnostic workup. Determination of the precise cause for male factor infertility is important for the followings:
understanding the mechanisms of disease which may open frontiers for disease prevention and treatment,
better clinical management decisions with the aim to find out optimal treatment methods for correctable conditions and to avoid unnecessary interventions,
assessment of potential health risks to offspring and
assessment of potential comorbidities to the patients (Fig. 1b).
It is highly likely that a substantial fraction of current idiopathic cases is caused by unknown genetic factors as thousands of genes contribute to male gonadal development, testicular function and the complex and highly coordinated process of spermatogenesis. There are also an increasing number of patients who are either symptomatic or asymptomatic carriers of pathogenic genetic defects that must be considered in patient management. Undiagnosed paternal genetic defects (e.g. pathogenic variants in CFTR or pleiotropic developmental genes, see below) may be transmitted to the offspring and in the worst case cause severe and untreatable disease.6–8 When considering assisted reproductive technologies (ARTs), genetic testing results may assist in predicting success of sperm retrieval, oocyte fertilization, implantation of the embryo and probability of the pregnancy progression until term. The benefit from molecular diagnostics reaches beyond the management and counselling of the infertile couple as testing for the identified genetic cause can be offered also to extended family members.
The accumulated data have shown that quantitative disturbances of spermatogenesis may serve as a biomarker for male overall future health. Over 50% of infertile men suffer from one or more chronic diseases or major general health conditions.4 A recent meta-analysis clearly indicated that infertile men have a higher risk of earlier death.9 Moreover, the risk of earlier mortality increases with the severity of spermatogenic impairment and reduction in semen quality. Precise mechanisms of this correlation observed in epidemiological analyses are not yet known; however, the contribution of pleiotropic genetic risk factors is an attractive and well-justified hypothesis. It is well known that men have a significantly lower number of visits to their primary care doctors and diagnostic services than women. Infertile men are evaluated often at the age of 20–40 years, which opens new frontiers for advanced molecular diagnostic approaches and personalized targeted medical services also for general male health conditions.
Areas of agreement—a broad spectrum of genetic defects linked to male infertility
Known causes of male infertility assessed in current clinical practice
Well-established genetic causes explain up to 10% of male infertility, including gross chromosomal aberrations, biallelic pathogenic variants in the CFTR gene and Y chromosomal microdeletions (Fig. 1a and c).4,5 For each of these conditions, international guidelines have been developed and applied in the clinical management for over 20 years.1–3
The first genetic link to male infertility was established in 1959, showing that men with Klinefelter syndrome (KS) have an extra X-chromosome, resulting in a 47,XXY karyotype (Fig. 1c).10 KS is a prevalent condition (~1–2 men in 1000) and one of the leading genetic causes of male infertility (3–4% of cases).11 KS patients are typically characterized by small testicles, non-obstructive azoospermia (NOA) and high levels of FSH and LH hormones, termed as hypergonadotropic hypogonadism. Gross chromosomal abnormality in KS leads to increased morbidity, including osteoporosis, metabolic, cardiovascular, neurocognitive and psychosocial disturbances, as well as earlier mortality.12 This year, the European Academy of Andrology published the first guidelines to enable standardized management of KS patients across the lifespan.11 Patients with KS wishing to become a father have the ART option based on testicular sperm aspiration or surgical extraction combined with intracytoplasmic sperm injection (TESE-ICSI) to fertilize the oocyte. Spermatozoa can be found in ~30–60% of patients with KS and the reported live birth rate by ART is ~16%.
Apart from KS, additional up to 1.7% of male infertility patients carry autosomal abnormalities, such as translocations or inversions.3–5 The nature of infertility management depends on the exact chromosomal rearrangement and the degree of spermatogenic impairment. Preimplantation and prenatal genetic testing options may be used to prevent the transmission of the genetic defect to the offspring. For example, carriers of Robertsonian translocations have a high risk to conceive an offspring with aneuploidy, asymptomatic carriers of microdeletions or duplications linked to clinical syndromes may father a child developing a severe developmental disorder.
Another well-established genetic factor for male infertility is the occurrence of biallelic pathogenic variants in the CFTR gene causing congenital bilateral absence of the vas deference (CBAVD) and obstructive azoospermia (OA) (1–2% of infertile men).13,14 CFTR is characterized by high allelic heterogeneity and broad phenotypic expressivity of pathogenic variants ranging from classic cystic fibrosis (CF) with the lung and pancreatic phenotype to cases with only seminal duct obstruction.15 Proper genetic diagnosis is important as some men with the primary diagnosis of CFTR-related male reproductive disorders may also express mild CF symptoms, such as recurrent respiratory tract or pancreatic infections.14 Diagnostic yield of genetic tests of CBAVD/OA is high, >80% in bilateral and >30% in unilateral cases (Table 1).14,15 CBAVD/OA does not usually affect the process of spermatogenesis. Clinical management of most cases uses TESE-ICSI with reported live-birth rates ~18–36%.13 Genetic counselling and testing of the partner prior to conception is needed to prevent the birth of a child with CF.
Current knowledge of recurrent monogenic causes implicated in male infertilitya
| Congenital developmental defects | Qualitative or quantitative defects in spermatogenesis | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | CBAVD | 46 XY, DSD | CHH | MMAF | Macroz. | Globoz. | NOA | SO |
| Male prevalence | 0.1% | Up to 0.02% | <0.01% | <0.01% | <0.01% | <0.01% | ~1% | ~5% |
| Known genes (n) | 2 | ~30 major (>60 reported) | ~30 major (>60 reported) | ~20 | 1 | 5 | Up to ~50c | Candidates |
| Examples of confident disease genes (LP/P variants in ClinVar, n)b | CFTR (770), ADGRG2 (9) | AR (166), SOX9 (62), SRD5A2 (46), WT1 (53), NR5A1 (33), HSD3B2 (32), SRY (28), HSD17B3 (21), POR (19), CYP11A1 (13); deletions (WT1, DMRT regions) and | CHD7 (401), FGFR1 (94), ANOS1 (32), GNRHR (22), FGF8 (16), PROKR2 (8), PROK2 (7), TACR3 (10), AMH (6), KISS1R (5); deletions (ANOS1) | DNAH1 (30), CFAP43 (13), CFAP44 (6), DNAH2 (5), TTC29 (5), DNAH17 (4), CFAP65 (4), FSIP2 (4) | AURKC (6) | DPY19L2: hom. gene deletions (most cases), rare SNVs (5) | TEX11 (2), FANCM (4), TEX14 (3), MSH4 (3), STAG3 (9), MEIOB (3), SYCE1 (3); M1AP (2), KLHL10 (1); deletions (DMRT1, TEX11, SYCE1) | Reported LP/P variants in ZMYND15, FANCM, MSH4, M1AP, LHB, TDRD9, RPL10L, NR5A1, etc. |
| duplications (DAX1, WNT4) | Full AZFa, AZFb or AZFc deletionsd AZFc r2/r3 inv + del (12 cases reported)e | |||||||
| Inheritance mode | aut. rec., XL | aut. dom., aut. rec., XL | aut. dom., aut. rec., XL, di- & oligogenic | aut. rec., XL | aut. rec. | aut. rec. | Mostly aut. rec. or XL, less known aut. dom., oligogenic | |
| Disturbed functions | Ductal obstruction | Gonadal & genital development; steroidogenic pathways | GnRH neuron development & migration | Structure & motility of the flagellum | Cell division | Acrosomal function | Failed or impaired spermatogenesis; possible qualitative sperm defects in SO | |
| Diagnostic yield | >80% (bi) >30% (uni) | Up to 50% | ~50% | 30–60% | n.a. | ~60% | Mostly < 5% (See Table 2) | n.a. |
| Genetic testing approaches | Single gene sequencing | Targeted gene panel sequencing ES and in silico gene panel | Single gene sequencing | ES Deletion analysis | Combined approaches | |||
| Added value in infertility management | ART options | Early sex assignment; multidisciplinary clinical management | Hormone therapy; multidisciplinary management | Optimal clinical solutions for the couple to achieve parenthood; personalised long- term treatment and management; prognosis of the success of TESE-ICSI | ||||
| Other benefits of genetic diagnosis | Early management of potential comorbidities, such as cancer, chronic disease, and psychological support; assessment of severe congenital, reproductive and general health risks to the future offspring; genetic testing of family members for their reproductive decision-making | |||||||
| Congenital developmental defects | Qualitative or quantitative defects in spermatogenesis | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | CBAVD | 46 XY, DSD | CHH | MMAF | Macroz. | Globoz. | NOA | SO |
| Male prevalence | 0.1% | Up to 0.02% | <0.01% | <0.01% | <0.01% | <0.01% | ~1% | ~5% |
| Known genes (n) | 2 | ~30 major (>60 reported) | ~30 major (>60 reported) | ~20 | 1 | 5 | Up to ~50c | Candidates |
| Examples of confident disease genes (LP/P variants in ClinVar, n)b | CFTR (770), ADGRG2 (9) | AR (166), SOX9 (62), SRD5A2 (46), WT1 (53), NR5A1 (33), HSD3B2 (32), SRY (28), HSD17B3 (21), POR (19), CYP11A1 (13); deletions (WT1, DMRT regions) and | CHD7 (401), FGFR1 (94), ANOS1 (32), GNRHR (22), FGF8 (16), PROKR2 (8), PROK2 (7), TACR3 (10), AMH (6), KISS1R (5); deletions (ANOS1) | DNAH1 (30), CFAP43 (13), CFAP44 (6), DNAH2 (5), TTC29 (5), DNAH17 (4), CFAP65 (4), FSIP2 (4) | AURKC (6) | DPY19L2: hom. gene deletions (most cases), rare SNVs (5) | TEX11 (2), FANCM (4), TEX14 (3), MSH4 (3), STAG3 (9), MEIOB (3), SYCE1 (3); M1AP (2), KLHL10 (1); deletions (DMRT1, TEX11, SYCE1) | Reported LP/P variants in ZMYND15, FANCM, MSH4, M1AP, LHB, TDRD9, RPL10L, NR5A1, etc. |
| duplications (DAX1, WNT4) | Full AZFa, AZFb or AZFc deletionsd AZFc r2/r3 inv + del (12 cases reported)e | |||||||
| Inheritance mode | aut. rec., XL | aut. dom., aut. rec., XL | aut. dom., aut. rec., XL, di- & oligogenic | aut. rec., XL | aut. rec. | aut. rec. | Mostly aut. rec. or XL, less known aut. dom., oligogenic | |
| Disturbed functions | Ductal obstruction | Gonadal & genital development; steroidogenic pathways | GnRH neuron development & migration | Structure & motility of the flagellum | Cell division | Acrosomal function | Failed or impaired spermatogenesis; possible qualitative sperm defects in SO | |
| Diagnostic yield | >80% (bi) >30% (uni) | Up to 50% | ~50% | 30–60% | n.a. | ~60% | Mostly < 5% (See Table 2) | n.a. |
| Genetic testing approaches | Single gene sequencing | Targeted gene panel sequencing ES and in silico gene panel | Single gene sequencing | ES Deletion analysis | Combined approaches | |||
| Added value in infertility management | ART options | Early sex assignment; multidisciplinary clinical management | Hormone therapy; multidisciplinary management | Optimal clinical solutions for the couple to achieve parenthood; personalised long- term treatment and management; prognosis of the success of TESE-ICSI | ||||
| Other benefits of genetic diagnosis | Early management of potential comorbidities, such as cancer, chronic disease, and psychological support; assessment of severe congenital, reproductive and general health risks to the future offspring; genetic testing of family members for their reproductive decision-making | |||||||
aMore details of known genetic causes of male infertility sub-phenotypes are provided in recent reviews: CBAVD,14 CHH,20,21 DSD,22,23 teratozoozoospermia,24 including MMAF26 and globozoospermia,25 NOA.27
bAssessed April 2021; likely pathogenic or pathogenic (LP/P) nonsense, frameshift, splicing and missense variants linked to infertility phenotypes.
cOnly few genes detected with pathogenic variants in NOA patients in more than two studies; less than 30 genes identified in at least two studies.27
dRef.16
eRef.46
aut. dom., autosomal-dominant; aut. rec., autosomal recessive; AUA, American Urological Association; bi, bilateral; CBAVD, congenital absence of the vas deferens; DSD, disorder of sex development; EAU, European Association of Urology; Globoz., globozoospermia; hom, homozygous; Macroz., macrozoospermia; MMAF, multiple morphological abnormalities of the sperm flagella; n.a., not available; seq, sequencing; SNV, single nucleotide variant; SO, severe oligozoospermia; TESE-ICSI, intracytoplasmic sperm injection; uni, unilateral; XL, X-linked.
Current knowledge of recurrent monogenic causes implicated in male infertilitya
| Congenital developmental defects | Qualitative or quantitative defects in spermatogenesis | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | CBAVD | 46 XY, DSD | CHH | MMAF | Macroz. | Globoz. | NOA | SO |
| Male prevalence | 0.1% | Up to 0.02% | <0.01% | <0.01% | <0.01% | <0.01% | ~1% | ~5% |
| Known genes (n) | 2 | ~30 major (>60 reported) | ~30 major (>60 reported) | ~20 | 1 | 5 | Up to ~50c | Candidates |
| Examples of confident disease genes (LP/P variants in ClinVar, n)b | CFTR (770), ADGRG2 (9) | AR (166), SOX9 (62), SRD5A2 (46), WT1 (53), NR5A1 (33), HSD3B2 (32), SRY (28), HSD17B3 (21), POR (19), CYP11A1 (13); deletions (WT1, DMRT regions) and | CHD7 (401), FGFR1 (94), ANOS1 (32), GNRHR (22), FGF8 (16), PROKR2 (8), PROK2 (7), TACR3 (10), AMH (6), KISS1R (5); deletions (ANOS1) | DNAH1 (30), CFAP43 (13), CFAP44 (6), DNAH2 (5), TTC29 (5), DNAH17 (4), CFAP65 (4), FSIP2 (4) | AURKC (6) | DPY19L2: hom. gene deletions (most cases), rare SNVs (5) | TEX11 (2), FANCM (4), TEX14 (3), MSH4 (3), STAG3 (9), MEIOB (3), SYCE1 (3); M1AP (2), KLHL10 (1); deletions (DMRT1, TEX11, SYCE1) | Reported LP/P variants in ZMYND15, FANCM, MSH4, M1AP, LHB, TDRD9, RPL10L, NR5A1, etc. |
| duplications (DAX1, WNT4) | Full AZFa, AZFb or AZFc deletionsd AZFc r2/r3 inv + del (12 cases reported)e | |||||||
| Inheritance mode | aut. rec., XL | aut. dom., aut. rec., XL | aut. dom., aut. rec., XL, di- & oligogenic | aut. rec., XL | aut. rec. | aut. rec. | Mostly aut. rec. or XL, less known aut. dom., oligogenic | |
| Disturbed functions | Ductal obstruction | Gonadal & genital development; steroidogenic pathways | GnRH neuron development & migration | Structure & motility of the flagellum | Cell division | Acrosomal function | Failed or impaired spermatogenesis; possible qualitative sperm defects in SO | |
| Diagnostic yield | >80% (bi) >30% (uni) | Up to 50% | ~50% | 30–60% | n.a. | ~60% | Mostly < 5% (See Table 2) | n.a. |
| Genetic testing approaches | Single gene sequencing | Targeted gene panel sequencing ES and in silico gene panel | Single gene sequencing | ES Deletion analysis | Combined approaches | |||
| Added value in infertility management | ART options | Early sex assignment; multidisciplinary clinical management | Hormone therapy; multidisciplinary management | Optimal clinical solutions for the couple to achieve parenthood; personalised long- term treatment and management; prognosis of the success of TESE-ICSI | ||||
| Other benefits of genetic diagnosis | Early management of potential comorbidities, such as cancer, chronic disease, and psychological support; assessment of severe congenital, reproductive and general health risks to the future offspring; genetic testing of family members for their reproductive decision-making | |||||||
| Congenital developmental defects | Qualitative or quantitative defects in spermatogenesis | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | CBAVD | 46 XY, DSD | CHH | MMAF | Macroz. | Globoz. | NOA | SO |
| Male prevalence | 0.1% | Up to 0.02% | <0.01% | <0.01% | <0.01% | <0.01% | ~1% | ~5% |
| Known genes (n) | 2 | ~30 major (>60 reported) | ~30 major (>60 reported) | ~20 | 1 | 5 | Up to ~50c | Candidates |
| Examples of confident disease genes (LP/P variants in ClinVar, n)b | CFTR (770), ADGRG2 (9) | AR (166), SOX9 (62), SRD5A2 (46), WT1 (53), NR5A1 (33), HSD3B2 (32), SRY (28), HSD17B3 (21), POR (19), CYP11A1 (13); deletions (WT1, DMRT regions) and | CHD7 (401), FGFR1 (94), ANOS1 (32), GNRHR (22), FGF8 (16), PROKR2 (8), PROK2 (7), TACR3 (10), AMH (6), KISS1R (5); deletions (ANOS1) | DNAH1 (30), CFAP43 (13), CFAP44 (6), DNAH2 (5), TTC29 (5), DNAH17 (4), CFAP65 (4), FSIP2 (4) | AURKC (6) | DPY19L2: hom. gene deletions (most cases), rare SNVs (5) | TEX11 (2), FANCM (4), TEX14 (3), MSH4 (3), STAG3 (9), MEIOB (3), SYCE1 (3); M1AP (2), KLHL10 (1); deletions (DMRT1, TEX11, SYCE1) | Reported LP/P variants in ZMYND15, FANCM, MSH4, M1AP, LHB, TDRD9, RPL10L, NR5A1, etc. |
| duplications (DAX1, WNT4) | Full AZFa, AZFb or AZFc deletionsd AZFc r2/r3 inv + del (12 cases reported)e | |||||||
| Inheritance mode | aut. rec., XL | aut. dom., aut. rec., XL | aut. dom., aut. rec., XL, di- & oligogenic | aut. rec., XL | aut. rec. | aut. rec. | Mostly aut. rec. or XL, less known aut. dom., oligogenic | |
| Disturbed functions | Ductal obstruction | Gonadal & genital development; steroidogenic pathways | GnRH neuron development & migration | Structure & motility of the flagellum | Cell division | Acrosomal function | Failed or impaired spermatogenesis; possible qualitative sperm defects in SO | |
| Diagnostic yield | >80% (bi) >30% (uni) | Up to 50% | ~50% | 30–60% | n.a. | ~60% | Mostly < 5% (See Table 2) | n.a. |
| Genetic testing approaches | Single gene sequencing | Targeted gene panel sequencing ES and in silico gene panel | Single gene sequencing | ES Deletion analysis | Combined approaches | |||
| Added value in infertility management | ART options | Early sex assignment; multidisciplinary clinical management | Hormone therapy; multidisciplinary management | Optimal clinical solutions for the couple to achieve parenthood; personalised long- term treatment and management; prognosis of the success of TESE-ICSI | ||||
| Other benefits of genetic diagnosis | Early management of potential comorbidities, such as cancer, chronic disease, and psychological support; assessment of severe congenital, reproductive and general health risks to the future offspring; genetic testing of family members for their reproductive decision-making | |||||||
aMore details of known genetic causes of male infertility sub-phenotypes are provided in recent reviews: CBAVD,14 CHH,20,21 DSD,22,23 teratozoozoospermia,24 including MMAF26 and globozoospermia,25 NOA.27
bAssessed April 2021; likely pathogenic or pathogenic (LP/P) nonsense, frameshift, splicing and missense variants linked to infertility phenotypes.
cOnly few genes detected with pathogenic variants in NOA patients in more than two studies; less than 30 genes identified in at least two studies.27
dRef.16
eRef.46
aut. dom., autosomal-dominant; aut. rec., autosomal recessive; AUA, American Urological Association; bi, bilateral; CBAVD, congenital absence of the vas deferens; DSD, disorder of sex development; EAU, European Association of Urology; Globoz., globozoospermia; hom, homozygous; Macroz., macrozoospermia; MMAF, multiple morphological abnormalities of the sperm flagella; n.a., not available; seq, sequencing; SNV, single nucleotide variant; SO, severe oligozoospermia; TESE-ICSI, intracytoplasmic sperm injection; uni, unilateral; XL, X-linked.
The third currently practiced set of genetic tests target recurrent (de novo) microdeletions of the Y-chromosomal Azoospermia Factor a, b and c (AZFa-c) regions. These deletions are identified in 2–6% of male infertility cases, but 6–15% of NOA and cryptozoospermia patients.4,5,16 Most deletion carriers have no or very low amount of sperm.17 Testing for AZF deletions has been strongly recommended in the diagnostic workup for infertility patients with sperm concentration of <5 mln/ml.13,16 Presence of AZFa or AZFb microdeletions predict a very poor prognosis for sperm retrieval. TESE is recommended for men with AZFc deletions (success rate up to ~80% of cases), and on some occasions, low amounts of sperm can be present in their ejaculate facilitating the use of the conventional in vitro fertilization (IVF) approach. The couple must be alerted that their male offspring will inherit the Y-chromosomal abnormality and will likely be infertile.
Confidently established monogenic causes of male infertility
The first systematic review and clinical validity assessment of the monogenic causes of male infertility was published in 2019, reporting 78 genes linked to 92 phenotypes.18 This number has rapidly grown, and by 2021, already 104 genes have been confidently linked to 120 male infertility phenotypes and abnormal genitourinary development.19 However, majority of these findings concern rare sub-phenotypes of the infertile patients that are infrequently seen in everyday andrology practice. About half of the established male infertility genes are implicated in rare developmental defects of gonads, adrenal glands, hypothalamus, pituitary or vas deferens, and additional 20% or more are related to spermiogenesis and the ultrarare conditions of qualitative sperm defects affecting motility and morphology. Still, the gathered knowledge from these confident research outcomes paves the way in translating genetic findings related to congenital reproductive disorders into clinical practice for the patients’ benefit.
For 46,XY subjects with rare but well defined clinical conditions, such as congenital hypogonadotropic hypogonadism (CHH) or disorders of sex development (DSD), the diagnostic yield of testing monogenic causes is already ~40–50% (Table 1). The recommendations regarding the personalized treatment and management options of CHH and DSD patients are well outlined by multidisciplinary expert groups. CHH is a rare condition (1/30 000–86 000 men) due to the failure of gonadotropin-releasing hormone (GnRH) secretion, leading to delayed puberty and infertility.20,21 It can be associated with various other anomalies and the most frequent is an absent sense of smell, termed as Kallmann syndrome. Timely diagnosis and treatment will induce puberty, leading to improved sexual, bone, metabolic and psychological health. Importantly, ~10–20% of male cases exhibit a spontaneous recovery of their reproductive function and fertility can be induced with hormonal treatment in most patients. The term DSD (1/4500–5000 men) represents a diverse spectrum of clinical disorders and aetiologies, including defective androgen synthesis/action, partial (hypospadias and cryptorchidism) or complete gonadal dysgenesis.22,23 Among subjects with any sign of DSD, 75% have 46,XY karyotype and the most extreme form is complete sex reversal (46,XY females; ~1/16 000 women). Lifelong management of reproductive and overall health of DSD patients starts in early childhood and is based on an individualized care plan depending on the detailed phenotype and genetic finding.
Exome sequencing (ES) that became available 10 years ago opened the ‘hunt’ for monogenic causes of various other subtypes of male infertility. It has brought along breakthroughs in the genetics of ultrarare defects of sperm morphology (teratozoospermia) and motility (asthenozoospermia), typically presenting without major decrease in sperm numbers. Some qualitative abnormalities of sperm, such as globo- and macrozoospermia, are due to pathogenic variants in a single gene, DPY19L2 and AURKC, respectively (Table 1).24,25 In contrast, multiple morphological abnormalities of the sperm flagella (MMAF) is caused by defects in a diverse set of proteins localized in the sperm flagella.26 Upon careful clinical phenotyping, the current diagnostic yield of targeted gene analysis reaches already ~60%. Pathogenic variants in some genes (e.g. DNAH17) have been reported in globozoospermia as well as MMAF, suggesting possible shared aetiology. A possible management solution in these conditions is TESE-ICSI; however, multiple studies have shown frequent aneuploidy rates and low-quality sperm nuclei related to sperm flagellar defects.24 Thus, close attention should be paid to genetic counselling and clinical decision-making in MMAF cases.
Areas of controversy—a gap in the knowledge of monogenic causes of quantitative sperm defects
In contrast to rare infertility phenotypes, the progress in increasing the knowledge about genetic causes of quantitative spermatogenic failure, the most prevalent form of infertility, has been slower and less successful (Table 1). NOA, referring to complete lack of sperm, affects 1 in 100 men and currently, ~20% of cases remain idiopathic (Fig. 1a). Five in 100 men suffer from severe oligozoospermia (SO, sperm count < 5 mln/ml) and among these, over 70% are unexplained cases.4 The number of genes that have been confidently linked to NOA and SO is limited, despite a general belief that idiopathic cases of these conditions are largely of genetic origin.18,27 Therefore, a large proportion of men are not assigned a diagnostic cause of their low sperm count and a high number of couples are undergoing infertility management that is not explicitly evidence-based. What could be the possible reasons for failing to identify the genetic causes with current research strategies?
A specific challenge in uncovering monogenic forms of male infertility is the lack of large pedigrees with the segregating disease as most patients are the only affected subjects. Thus, the mode of inheritance cannot be straightforwardly detected, and genetic tools successfully used for other Mendelian phenotypes cannot be fully exploited. This complicates interpreting and making conclusions about the causative nature of the identified candidate genetic variants. New approaches and solutions must be looked for, such as confirmation of the genotype–phenotype link using animal models.28 However, some genes that are linked to male infertility phenotypes in murine models do not affect human spermatogenesis. For example, homozygous mice lacking the functional gene PRDM9, responsible for meiotic recombination, show azoospermia, decreased oocyte number and sterility in both sexes, but the described human knockouts are healthy and fertile.29 On the contrary, pathogenic variants in XRCC2 are linked to NOA in human,30 but mouse models present developmental defects and respiratory failure without affected reproduction.
Although most of the extreme infertility cases are expected to be sporadic, family anamnesis and genetic analysis of their immediate relatives assists in distinguishing between rare variants with and without potential effect on spermatogenesis, as well as identifying de novo causes of infertility. However, family planning is usually considered a highly private matter and having difficulties in achieving parenthood is typically not discussed among relatives. In many instances, the motivation and psychological readiness of either the patient or relatives restrict the recruitment, genetic and clinical assessment of the extended family members.
A large proportion of the reported monogenic forms of spermatogenic defects represent homozygous autosomal recessive pathogenic variants mapped in consanguineous families.27 A strength of these studies has been the availability of several family members for genetic analysis. However, the presence of long chromosomal regions that are homozygous in subjects from consanguineous marriages restricts confident definition of an identified variant as a monogenic cause for infertility. Pleiotropic effects of variants causing other phenotypic complications reported in these family-cases must be taken with reservation until confirmed by independent evidence. Also, particular genes or their combinations may carry ultra-rare variants found only in a single or a few consanguineous families worldwide and may seldom or even never be identified among infertility patients in outbred populations. Genetic ‘matchmaking’ is necessary to establish an explicit link between a particular monogenic defect and male infertility. Recent studies have innovatively combined the evidence gathered from family-based and sporadic cases in the identification of novel infertility-related genes.31,32
Growing points—clinical value of recent advances in monogenic causes of infertility
Expanded gene panels for clinically well-defined specific conditions
Clinicians managing infertile men are waiting for the translation of the most confident recent genetic findings from research to the practice along with internationally developed and applicable guidelines. Currently, none of the clinical guidelines include mutational analysis of genes linked to DSD, CHH, quantitative or qualitative defects of spermatogenesis.2,13 The rationale to expand genetic testing with monogenic forms of male infertility aims to confirm an explicit congenital cause and a sub-phenotype of the condition and to provide maximal evidence-based counselling, and the most optimal management. The options to achieve fatherhood range from non-invasive solutions including hormonal therapies, supportive care or life-style changes to various invasive approaches, such as conventional IVF, surgical correction of urogenital tract or TESE-ICSI. Non-invasive tools should be preferred whenever there is enough evidence to predict a successful outcome.13 Highly invasive TESE-ICSI used for severe male factor infertility cases has lower delivery rate compared with conventional IVF (~20 vs. ~30%).7 When opting for the invasive, time-consuming and costly ICSI procedure using testicular or epididymal sperm, specific genetic diagnosis will allow confident prognosis of fatherhood probability. In addition to potential health complications of TESE to the male partner and ART-related applications to the female partner, there are increased risks of long-term health and developmental outcomes in children conceived with ICSI, such as congenital malformations, chromosomal and epigenetic abnormalities, autism and neurological conditions, subfertility and childhood cancer.6,7 These may arise from the nature of the procedure itself but may also be due to paternal pleiotropic genetic variants that predispose to both, infertility and developmental or other genetic disorders.8 Thus, genetic diagnosis and respective counselling is critical to inform about the possible health risks of the future offspring. For example, DNAH1 implicated in MMAF is also linked to primary ciliary dyskinesia (PCD); WT1 pathogenic variants cause gonadal and renal maldevelopment but also childhood kidney cancer; an azoospermic man due to a pathogenic variant in the NR5A1 gene may father a child with sex reversal when infertility management uses testicular or epididymal sperm for ICSI.
For the rare conditions—DSD, CHH and structural defects of the sperm, the accumulated knowledge is already sufficient to develop well-standardized targeted gene panels that would further promote systematic collection and recording of clinical outcomes of genotype-driven management schemes. Regarding genetic testing in men with qualitative sperm defects, there is an urgent need to develop internationally standardized tests and subsequent clinical recommendations. Insufficient publications on genotype-stratified clinical data on the success of ICSI are the current main limitation towards this goal.26 In cases of CBAVD/OA, future recommendations should also include testing for the recently discovered recurrently mutated gene, ADGRG2.33
Strategies for the clinical ES of infertile men
There is a general agreement in both, basic research and clinical community, that cases with idiopathic severe quantitative spermatogenic failure are likely to be caused by yet unknown genetic factors and their identification is crucial in improving patient management. However, for this major form of male infertility, no ‘recurrently’ mutated key genes have been identified that can be straightforwardly included into genetic testing of patients to increase the diagnostic yield. Although several studies have attempted to establish diagnostic gene panels for the known NOA and SO genes, the confident diagnostic yield is modest, mostly below 5% (across 10 published studies: range 0–13.6%, mean 3.4%, median 2.2%; Table 2). Recent studies have shown that usage of ES for the analysis of previously reported genes combined with the discovery of novel infertility genes increases the yield manifold.34–36
Diagnostic yield of sequencing gene panels designed for the reported monogenic causes of male infertility
| Study | Design of studies analysing gene panels of reported male infertility loci | Re-assessed pathogenicity of the published disease-linked genotypes (number of cases shown, if n > 1)a | Diagnostic yield | |||
|---|---|---|---|---|---|---|
| No of analysed genes | Cases (n; phenotype) | Likely pathogenic/Pathogenic (LP/P) | Uncertain significance | Likely benign/Benign | Cases with LP/P (%) | |
| Oud et al. 201752 | 107 | 1112; NOA & SO | CFTR (5), SYCP3 | 0 | 0 | 6 of 1112 (0.54%) |
| Araujo et al. 202053 | 37 | 16; NOA | 0 | KLHL10, digenic SYCE1L*/REC8* | KLHL10, TEX15, TEX14, DMRT1, DNMT3Bb,* | 0 of 16 (0%) |
| Rocca et al. 202039 | 9 | 174; NOA & SO | AR (XL; 2, shared variant), NR5A1 (2) | KLHL10 (2, shared variant), SEPT12 (3), NR5A1, SYCP3 (2) | 0 | 4 of 174 (2.3%) |
| Cannarella et al. 202040 | 110 | 22; PCD, CHH, NOA & SO | CCDC39** (PCD case)c, NR5A1 (2) | CHD7 (CHH case) | TEX11 (XL) | 3 of 22 (13.6%) |
| Chen et al. 202036 | 36 | 314; NOA & SO | AR (XL; 2), TEX11 (XL; 2), DMC1, MEI1 | WNK3 (XL; 2), HAUS7 (XL), TEX14 | 0 | 6 of 314 (1.9%) |
| Alhathal et al. 202035 | ES and in silico look upd: Panel 1—OMIM: male infertility 2—Reported genes not in OMIM | 285; NOA & SO; study included 127 (44.6%) family cases | 1 − DNAH1 (2), SLC26A8, CFAP44, CFTR, DNAH5**, XRCC2, ZMYND15 2—CCDC155 (3, shared variant), DZIP1 | 1 − DNAAF2**, HYDIN**, SYCP2, TTC21A, NANOS1, TEX11 (XL), USP9Y (YL) 2—DNAH6, SPAG17, TDRD6, FAM47C (XL) | 0 | Panel 1 + 2: 12 of 285 (4.2%) |
| Precone et al. 202154 | 175 | 12; NOA, SO, OAT & CHH | 0 | DNAH11**, DNAH10, CATSPER2, AMELY (YL)* | CCDC40**, ADCY10e | 0 of 12 (0%) |
| Okutman et al. 202155 | 36 | 79; NOA, SO, OAT & MMAF | KLHL10, AURKC, CFAP43, DNAH1, GALNTL5 | 0 | 0 | 5 of 79 (6.3%) |
| An et al. 202141 | Panel 1 − TEX11, AR, KLHL10, NR5A1, SYCP3 Panel 2—panel 1 & nine additional genes | 668; NOA Panel 1 − 189 Panel 2 − 479 | NR5A1 (4), TEX11 (XL; 2), TEX14 (3), KLHL10, SYCE1, ZMYND15 | KLHL10 (2), AR (XL; 2), TEX11 (XL; 2) | 0 | 14 of 688 (2.1%) |
| Batiha et al. 202156 | 134 | 69; azoosp. | CFTR (2), USP9Y (YL) | MCM8f,*, KDM5D (YL)*, MAST2*, MEIOB, UTP14C, DNAH6 | 0 | 3 of 69 (4.3%) |
| Study | Design of studies analysing gene panels of reported male infertility loci | Re-assessed pathogenicity of the published disease-linked genotypes (number of cases shown, if n > 1)a | Diagnostic yield | |||
|---|---|---|---|---|---|---|
| No of analysed genes | Cases (n; phenotype) | Likely pathogenic/Pathogenic (LP/P) | Uncertain significance | Likely benign/Benign | Cases with LP/P (%) | |
| Oud et al. 201752 | 107 | 1112; NOA & SO | CFTR (5), SYCP3 | 0 | 0 | 6 of 1112 (0.54%) |
| Araujo et al. 202053 | 37 | 16; NOA | 0 | KLHL10, digenic SYCE1L*/REC8* | KLHL10, TEX15, TEX14, DMRT1, DNMT3Bb,* | 0 of 16 (0%) |
| Rocca et al. 202039 | 9 | 174; NOA & SO | AR (XL; 2, shared variant), NR5A1 (2) | KLHL10 (2, shared variant), SEPT12 (3), NR5A1, SYCP3 (2) | 0 | 4 of 174 (2.3%) |
| Cannarella et al. 202040 | 110 | 22; PCD, CHH, NOA & SO | CCDC39** (PCD case)c, NR5A1 (2) | CHD7 (CHH case) | TEX11 (XL) | 3 of 22 (13.6%) |
| Chen et al. 202036 | 36 | 314; NOA & SO | AR (XL; 2), TEX11 (XL; 2), DMC1, MEI1 | WNK3 (XL; 2), HAUS7 (XL), TEX14 | 0 | 6 of 314 (1.9%) |
| Alhathal et al. 202035 | ES and in silico look upd: Panel 1—OMIM: male infertility 2—Reported genes not in OMIM | 285; NOA & SO; study included 127 (44.6%) family cases | 1 − DNAH1 (2), SLC26A8, CFAP44, CFTR, DNAH5**, XRCC2, ZMYND15 2—CCDC155 (3, shared variant), DZIP1 | 1 − DNAAF2**, HYDIN**, SYCP2, TTC21A, NANOS1, TEX11 (XL), USP9Y (YL) 2—DNAH6, SPAG17, TDRD6, FAM47C (XL) | 0 | Panel 1 + 2: 12 of 285 (4.2%) |
| Precone et al. 202154 | 175 | 12; NOA, SO, OAT & CHH | 0 | DNAH11**, DNAH10, CATSPER2, AMELY (YL)* | CCDC40**, ADCY10e | 0 of 12 (0%) |
| Okutman et al. 202155 | 36 | 79; NOA, SO, OAT & MMAF | KLHL10, AURKC, CFAP43, DNAH1, GALNTL5 | 0 | 0 | 5 of 79 (6.3%) |
| An et al. 202141 | Panel 1 − TEX11, AR, KLHL10, NR5A1, SYCP3 Panel 2—panel 1 & nine additional genes | 668; NOA Panel 1 − 189 Panel 2 − 479 | NR5A1 (4), TEX11 (XL; 2), TEX14 (3), KLHL10, SYCE1, ZMYND15 | KLHL10 (2), AR (XL; 2), TEX11 (XL; 2) | 0 | 14 of 688 (2.1%) |
| Batiha et al. 202156 | 134 | 69; azoosp. | CFTR (2), USP9Y (YL) | MCM8f,*, KDM5D (YL)*, MAST2*, MEIOB, UTP14C, DNAH6 | 0 | 3 of 69 (4.3%) |
aMale infertility linked variants reported in the literature were re-assessed and re-classified for their pathogenicity in August 2021 based on the ACMG guidelines.37 In this pathogenicity re-assessment only genotypes relevant to the inheritance mode (based on the OMIM database) were considered—rare homo- or compound heterozygous variant carriers for autosomal recessive genes, heterozygotes for autosomal dominant and hemizygotes for X-linked (XL) or Y-linked (YL) genes.
bHeterozygous LP/P variant in a gene linked to a distinct recessive phenotype (immunodeficiency-centromeric instability-facial anomalies syndrome 1; MIM: 242860).
cCompound heterozygote for LP/P and variant of uncertain significance.
dThe study used ES dataset with dual purposes—discovery of novel male infertility linked genes and targeted analysis of previously reported loci; this table reports only the outcome of targeted candidate gene analysis; exact number and list of assessed genes in these gene panels were not available.
eHeterozygous LP/P variant reported in a gene linked to a distinct dominant phenotype (absorptive hypercalciuria; MIM: 143870); only homozygosity of pathogenic variants has been additionally linked to severe recessive asthenozoospermia.57
fLinked to premature ovarian failure (MIM: 612885).
*No publications reporting a causal relationship between LP/P missense/loss-of-function variants in this gene and male infertility.
**Primary linked phenotype is PCD.
azoosp., azoospermia; MMAF, Multiple morphological abnormalities of the sperm flagella; OAT, oligoasthenozoospermia; SF, spermatogenic failure; SO, severe oligozoospermia.
Diagnostic yield of sequencing gene panels designed for the reported monogenic causes of male infertility
| Study | Design of studies analysing gene panels of reported male infertility loci | Re-assessed pathogenicity of the published disease-linked genotypes (number of cases shown, if n > 1)a | Diagnostic yield | |||
|---|---|---|---|---|---|---|
| No of analysed genes | Cases (n; phenotype) | Likely pathogenic/Pathogenic (LP/P) | Uncertain significance | Likely benign/Benign | Cases with LP/P (%) | |
| Oud et al. 201752 | 107 | 1112; NOA & SO | CFTR (5), SYCP3 | 0 | 0 | 6 of 1112 (0.54%) |
| Araujo et al. 202053 | 37 | 16; NOA | 0 | KLHL10, digenic SYCE1L*/REC8* | KLHL10, TEX15, TEX14, DMRT1, DNMT3Bb,* | 0 of 16 (0%) |
| Rocca et al. 202039 | 9 | 174; NOA & SO | AR (XL; 2, shared variant), NR5A1 (2) | KLHL10 (2, shared variant), SEPT12 (3), NR5A1, SYCP3 (2) | 0 | 4 of 174 (2.3%) |
| Cannarella et al. 202040 | 110 | 22; PCD, CHH, NOA & SO | CCDC39** (PCD case)c, NR5A1 (2) | CHD7 (CHH case) | TEX11 (XL) | 3 of 22 (13.6%) |
| Chen et al. 202036 | 36 | 314; NOA & SO | AR (XL; 2), TEX11 (XL; 2), DMC1, MEI1 | WNK3 (XL; 2), HAUS7 (XL), TEX14 | 0 | 6 of 314 (1.9%) |
| Alhathal et al. 202035 | ES and in silico look upd: Panel 1—OMIM: male infertility 2—Reported genes not in OMIM | 285; NOA & SO; study included 127 (44.6%) family cases | 1 − DNAH1 (2), SLC26A8, CFAP44, CFTR, DNAH5**, XRCC2, ZMYND15 2—CCDC155 (3, shared variant), DZIP1 | 1 − DNAAF2**, HYDIN**, SYCP2, TTC21A, NANOS1, TEX11 (XL), USP9Y (YL) 2—DNAH6, SPAG17, TDRD6, FAM47C (XL) | 0 | Panel 1 + 2: 12 of 285 (4.2%) |
| Precone et al. 202154 | 175 | 12; NOA, SO, OAT & CHH | 0 | DNAH11**, DNAH10, CATSPER2, AMELY (YL)* | CCDC40**, ADCY10e | 0 of 12 (0%) |
| Okutman et al. 202155 | 36 | 79; NOA, SO, OAT & MMAF | KLHL10, AURKC, CFAP43, DNAH1, GALNTL5 | 0 | 0 | 5 of 79 (6.3%) |
| An et al. 202141 | Panel 1 − TEX11, AR, KLHL10, NR5A1, SYCP3 Panel 2—panel 1 & nine additional genes | 668; NOA Panel 1 − 189 Panel 2 − 479 | NR5A1 (4), TEX11 (XL; 2), TEX14 (3), KLHL10, SYCE1, ZMYND15 | KLHL10 (2), AR (XL; 2), TEX11 (XL; 2) | 0 | 14 of 688 (2.1%) |
| Batiha et al. 202156 | 134 | 69; azoosp. | CFTR (2), USP9Y (YL) | MCM8f,*, KDM5D (YL)*, MAST2*, MEIOB, UTP14C, DNAH6 | 0 | 3 of 69 (4.3%) |
| Study | Design of studies analysing gene panels of reported male infertility loci | Re-assessed pathogenicity of the published disease-linked genotypes (number of cases shown, if n > 1)a | Diagnostic yield | |||
|---|---|---|---|---|---|---|
| No of analysed genes | Cases (n; phenotype) | Likely pathogenic/Pathogenic (LP/P) | Uncertain significance | Likely benign/Benign | Cases with LP/P (%) | |
| Oud et al. 201752 | 107 | 1112; NOA & SO | CFTR (5), SYCP3 | 0 | 0 | 6 of 1112 (0.54%) |
| Araujo et al. 202053 | 37 | 16; NOA | 0 | KLHL10, digenic SYCE1L*/REC8* | KLHL10, TEX15, TEX14, DMRT1, DNMT3Bb,* | 0 of 16 (0%) |
| Rocca et al. 202039 | 9 | 174; NOA & SO | AR (XL; 2, shared variant), NR5A1 (2) | KLHL10 (2, shared variant), SEPT12 (3), NR5A1, SYCP3 (2) | 0 | 4 of 174 (2.3%) |
| Cannarella et al. 202040 | 110 | 22; PCD, CHH, NOA & SO | CCDC39** (PCD case)c, NR5A1 (2) | CHD7 (CHH case) | TEX11 (XL) | 3 of 22 (13.6%) |
| Chen et al. 202036 | 36 | 314; NOA & SO | AR (XL; 2), TEX11 (XL; 2), DMC1, MEI1 | WNK3 (XL; 2), HAUS7 (XL), TEX14 | 0 | 6 of 314 (1.9%) |
| Alhathal et al. 202035 | ES and in silico look upd: Panel 1—OMIM: male infertility 2—Reported genes not in OMIM | 285; NOA & SO; study included 127 (44.6%) family cases | 1 − DNAH1 (2), SLC26A8, CFAP44, CFTR, DNAH5**, XRCC2, ZMYND15 2—CCDC155 (3, shared variant), DZIP1 | 1 − DNAAF2**, HYDIN**, SYCP2, TTC21A, NANOS1, TEX11 (XL), USP9Y (YL) 2—DNAH6, SPAG17, TDRD6, FAM47C (XL) | 0 | Panel 1 + 2: 12 of 285 (4.2%) |
| Precone et al. 202154 | 175 | 12; NOA, SO, OAT & CHH | 0 | DNAH11**, DNAH10, CATSPER2, AMELY (YL)* | CCDC40**, ADCY10e | 0 of 12 (0%) |
| Okutman et al. 202155 | 36 | 79; NOA, SO, OAT & MMAF | KLHL10, AURKC, CFAP43, DNAH1, GALNTL5 | 0 | 0 | 5 of 79 (6.3%) |
| An et al. 202141 | Panel 1 − TEX11, AR, KLHL10, NR5A1, SYCP3 Panel 2—panel 1 & nine additional genes | 668; NOA Panel 1 − 189 Panel 2 − 479 | NR5A1 (4), TEX11 (XL; 2), TEX14 (3), KLHL10, SYCE1, ZMYND15 | KLHL10 (2), AR (XL; 2), TEX11 (XL; 2) | 0 | 14 of 688 (2.1%) |
| Batiha et al. 202156 | 134 | 69; azoosp. | CFTR (2), USP9Y (YL) | MCM8f,*, KDM5D (YL)*, MAST2*, MEIOB, UTP14C, DNAH6 | 0 | 3 of 69 (4.3%) |
aMale infertility linked variants reported in the literature were re-assessed and re-classified for their pathogenicity in August 2021 based on the ACMG guidelines.37 In this pathogenicity re-assessment only genotypes relevant to the inheritance mode (based on the OMIM database) were considered—rare homo- or compound heterozygous variant carriers for autosomal recessive genes, heterozygotes for autosomal dominant and hemizygotes for X-linked (XL) or Y-linked (YL) genes.
bHeterozygous LP/P variant in a gene linked to a distinct recessive phenotype (immunodeficiency-centromeric instability-facial anomalies syndrome 1; MIM: 242860).
cCompound heterozygote for LP/P and variant of uncertain significance.
dThe study used ES dataset with dual purposes—discovery of novel male infertility linked genes and targeted analysis of previously reported loci; this table reports only the outcome of targeted candidate gene analysis; exact number and list of assessed genes in these gene panels were not available.
eHeterozygous LP/P variant reported in a gene linked to a distinct dominant phenotype (absorptive hypercalciuria; MIM: 143870); only homozygosity of pathogenic variants has been additionally linked to severe recessive asthenozoospermia.57
fLinked to premature ovarian failure (MIM: 612885).
*No publications reporting a causal relationship between LP/P missense/loss-of-function variants in this gene and male infertility.
**Primary linked phenotype is PCD.
azoosp., azoospermia; MMAF, Multiple morphological abnormalities of the sperm flagella; OAT, oligoasthenozoospermia; SF, spermatogenic failure; SO, severe oligozoospermia.
Given the large number of genes implicated in spermatogenesis and testicular function, a high genetic heterogeneity is expected. An optimal molecular diagnostic tool would be sequencing exomes or in perspective, even genomes. Hand in hand, the development of data analysis and interpretation pipeline customized for male reproductive medicine must be undertaken (termed as ‘andro-exome pipeline’). To achieve this objective, several critical aspects must be fulfilled. Firstly, systematic research standards and quality criteria for reporting novel infertility related genes are required. Not all published claims correspond to today’s international guidelines established for reporting genetic variants,37 creating uncertainties in their clinical diagnostic value. It must be acknowledged that pathogenic variants in several genes linked to male infertility have variable phenotypic effect on reproductive and non-reproductive organs, modulated by other genetic and non-genetic contributors. Typical examples are biallelic CFTR variants with a broad phenotypic spectrum from CBAVD/OA to classic untreatable CF, or heterozygous pathogenic variants in NR5A1 initially described in DSD patients but now reported in patients presenting only NOA/SO, and asymptomatic cases.27,38–41 Secondly, clear clinical guidelines must be developed and annually updated to standardize the utilization of ‘andro-exome pipeline’ in the clinical practice worldwide.
In addition, cascade genetic testing of identified pathogenic variants in single genes should be offered to family members of male infertility patients, including assessment of female relatives. A large proportion of genes causing male conditions have also been implicated in female reproductive disorders and infertility, e.g. gonadal dysgenesis (DSD genes), amenorrhea (CHH genes) and premature ovarian insufficiency (e.g. NOA genes). Whereas gonadal ambiguities are usually documented at birth and amenorrhea in puberty, POI can also manifest with age and may not be present in a severe form in young women. Attention must also be paid to the counselling of asymptomatic carriers of variants causing dominant conditions with reduced penetrance or female carriers of pathogenic variants related to X-linked conditions.
Finally, introduction of ‘andro-exomes’ would also enable to detect secondary genetic findings42 and adjust the patient’s personal health management scheme, switching to genome-informed medical care. It is confidently established that 2–3% of subjects are carriers of clinically actionable variants in genes causing adult-onset inherited diseases that are preventable or manageable upon early detection.43,44
Areas timely for developing research—diversifying the scope of basic and clinical research
The key aim in basic research of male infertility genetics is to broaden the palette of approaches to uncover the ‘hidden’ genetic factors (Fig. 2a and b). One possible scenario is di−/oligogenic inheritance that is determined by the combination of rare pathogenic variants with reduced individual penetrance or variable expressivity. Oligogenic contributors to the rare CHH phenotype have been convincingly shown and oligogenic causes have been estimated for up to 15% of patients.20 Also for DSD cases, the observed variable penetrance and broad phenotypic variability among patients carrying the same pathogenic variant has been explained by modulatory variants in other loci.38,45
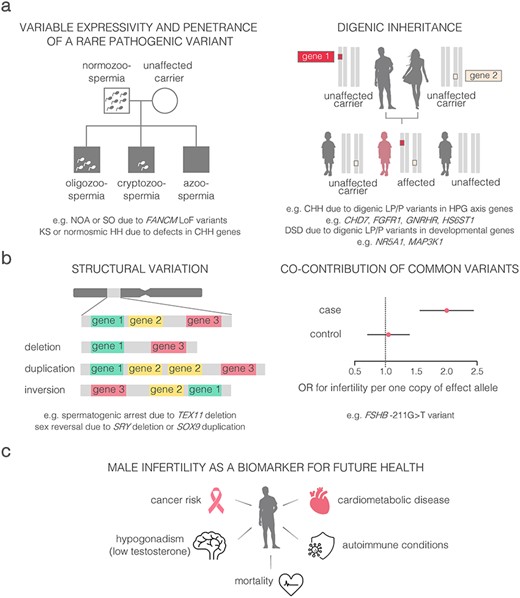
Perspectives to uncover ‘hidden’ genetics of male infertility and the link to general health. (a) Variable penetrance and expressivity of rare pathogenic variants. (b) Alternative genetic factors contributing to male infertility; (c) shared genetic landscape of male infertility and general health. CHH, congenital hypogonadotropic hypogonadism; DSD, disorders of sex development; HH, hypogonadotropic hypogonadism; HPG, hypothalamic–pituitary-gonadal; KS, Klinefelter syndrome; LoF, loss-of-function; LP, likely pathogenic; NOA, non-obstructive azoospermia; P, pathogenic; SO, severe oligozoospermia.
Altered dosage of single genes or larger chromosomal regions (e.g. DPY19L2, WT1, ANOS1, TEX11, DMRT1) represents a further perspective, but less explored contributor to infertility (Table 1). In addition to the known extreme effect of the full Y-chromosomal AZFa-c deletions on fertility potential, also the carriers of AZFc partial deletions are predisposed to spermatogenic failure. However, this risk is dependent on the demographics and Y-chromosomal lineage of the subject.17 A recent study in 2324 Estonian men uncovered a novel large-scale Y-chromosomal rearrangement increasing the risk for severe spermatogenic failure nearly 9-fold.46 Notably, this risk variant was mapped to a specific Y-chromosomal lineage R1a1-M458 that has >20% frequency in several Central-European populations. This finding re-draws the attention to the unique genetics, structure and evolutionary dynamics of the Y-chromosome, promoting future research to uncover and fine-map additional Y-lineage specific structural variants.
There is almost no information concerning common variants implicated in polygenic and multifactorial inheritance of male infertility. Unlike other phenotypes, the era of genome-wide association studies for key andrological parameters is still ahead. One of the few confidently established modulators is a promoter variant, FSHB-211 G > T, discovered by our team.47 Homozygous men for the minor allele (TT-genotype) exhibit significantly lower FSH and testes volume. Both male and female TT-homozygotes have more than 2-fold increased risk to infertility.48,49
Epidemiological studies have shown male factor infertility as a biomarker for future health, given that poor semen parameters are associated with an increased risk of hypogonadism, cardiometabolic disease, cancer and even earlier mortality.9,50 The potential comorbidities of known genetic factors, such as KS and the carriership of CFTR pathogenic variants, are long known, and respective management guidelines are available.11–14 It is critical to understand how the recently identified diverse palette of genetic contributors to infertility link to general health concerns across lifespan (Fig. 2c). For example, NOA patients exhibit increased risk to various cancers, possibly due to genetic defects in pathways regulating cell cycle and genomic integrity. Several genes responsible for DNA repair (e.g. FANCM, XRCC2, MSH4) have been already identified carrying pathogenic variants predisposing to both, infertility as well as familial cancer in men and women.27,30,31,51 Taken together, an important future direction is multidisciplinary clinical research and patient management, especially for the syndromic infertility cases with high risk to other health concerns and chronic diseases.
Glossary
| Aneuploidy | Abnormal number of chromosomes in a cell. |
| Aspermia | Complete lack of semen with ejaculation. |
| Assisted reproductive technology (ART) | Infertility treatment that involves removal of oocytes from a woman’s body, fertilizing them with sperm in the laboratory and transferring the resulting embryos back in the woman’s body. |
| Azoospermia | Complete lack of sperm in the ejaculate. |
| Biallelic | Variants detected in both gene alleles located on homologous chromosomes. |
| Clinically actionable genetic variant | Likely pathogenic or pathogenic variants in genes associated with diseases that are moderately to highly penetrant. |
| ClinVar database | ClinVar is a public archive with free access to reports on the relationships between human genetic variations and phenotypes, with supporting evidence. |
| Congenital bilateral absence of the vas deferens (CBAVD) | Several parts of the reproductive tract (the vas deferens, most of the epididymis and seminal vesicles) are missing in both sides of the body from birth. |
| Congenital hypogonadotropic hypogonadism (CHH) | Congenital defects in the hypothalamic–pituitary-gonadal axis leading to impaired central hormonal regulation of testis function. Frequently correctable. |
| Cryptozoospermia | Spermatozoa detectable only after centrifugation of the semen sample. |
| Di- and oligogenic inheritance | Genetic disease caused by the combined effect of two (or a few) clinically actionable variants. |
| Diagnostic gene panel | List of genes analysed for clinically actionable genetic variants using sequencing or genotyping in patients presenting the relevant phenotype. |
| Diagnostic yield | The likelihood that a test will result in the diagnostic cause of the disease. |
| Disorders of sex development (DSD) | Any problem where the genitalia are atypical in relation to the gonads or chromosomes (46,XY DSD or 46,XX DSD). |
| Exome sequencing | Sequencing of the coding part of the genome (1–2%). |
| Globozoospermia | Sperm cells with round head and no acrosome. |
| Isolated infertility | Infertility without any other apparent health problems. |
| Likely pathogenic/pathogenic variant (LP/P) | There is a high likelihood (greater than 90% certainty) that a variant is disease-causing/Causative for a disease. |
| Loss-of-function variant (LoF) | Genetic variant leading to a truncated protein, including nonsense (STOP), frameshift (fs) and splicing variants. |
| Macrozoospermia | Large-headed multiflagellar spermatozoa. |
| Multiple morphological abnormalities of the sperm flagella (MMAF) | Sperm with a mosaic of various flagellar abnormalities, such as absent, short, bent, coiled, and irregular flagella. |
| Non-obstructive azoospermia (NOA) | Lack of sperm in the ejaculate caused by congenital or acquired testicular disorders or primary spermatogenic failure. Usually uncorrectable. |
| Aneuploidy | Abnormal number of chromosomes in a cell. |
| Aspermia | Complete lack of semen with ejaculation. |
| Assisted reproductive technology (ART) | Infertility treatment that involves removal of oocytes from a woman’s body, fertilizing them with sperm in the laboratory and transferring the resulting embryos back in the woman’s body. |
| Azoospermia | Complete lack of sperm in the ejaculate. |
| Biallelic | Variants detected in both gene alleles located on homologous chromosomes. |
| Clinically actionable genetic variant | Likely pathogenic or pathogenic variants in genes associated with diseases that are moderately to highly penetrant. |
| ClinVar database | ClinVar is a public archive with free access to reports on the relationships between human genetic variations and phenotypes, with supporting evidence. |
| Congenital bilateral absence of the vas deferens (CBAVD) | Several parts of the reproductive tract (the vas deferens, most of the epididymis and seminal vesicles) are missing in both sides of the body from birth. |
| Congenital hypogonadotropic hypogonadism (CHH) | Congenital defects in the hypothalamic–pituitary-gonadal axis leading to impaired central hormonal regulation of testis function. Frequently correctable. |
| Cryptozoospermia | Spermatozoa detectable only after centrifugation of the semen sample. |
| Di- and oligogenic inheritance | Genetic disease caused by the combined effect of two (or a few) clinically actionable variants. |
| Diagnostic gene panel | List of genes analysed for clinically actionable genetic variants using sequencing or genotyping in patients presenting the relevant phenotype. |
| Diagnostic yield | The likelihood that a test will result in the diagnostic cause of the disease. |
| Disorders of sex development (DSD) | Any problem where the genitalia are atypical in relation to the gonads or chromosomes (46,XY DSD or 46,XX DSD). |
| Exome sequencing | Sequencing of the coding part of the genome (1–2%). |
| Globozoospermia | Sperm cells with round head and no acrosome. |
| Isolated infertility | Infertility without any other apparent health problems. |
| Likely pathogenic/pathogenic variant (LP/P) | There is a high likelihood (greater than 90% certainty) that a variant is disease-causing/Causative for a disease. |
| Loss-of-function variant (LoF) | Genetic variant leading to a truncated protein, including nonsense (STOP), frameshift (fs) and splicing variants. |
| Macrozoospermia | Large-headed multiflagellar spermatozoa. |
| Multiple morphological abnormalities of the sperm flagella (MMAF) | Sperm with a mosaic of various flagellar abnormalities, such as absent, short, bent, coiled, and irregular flagella. |
| Non-obstructive azoospermia (NOA) | Lack of sperm in the ejaculate caused by congenital or acquired testicular disorders or primary spermatogenic failure. Usually uncorrectable. |
Glossary
| Aneuploidy | Abnormal number of chromosomes in a cell. |
| Aspermia | Complete lack of semen with ejaculation. |
| Assisted reproductive technology (ART) | Infertility treatment that involves removal of oocytes from a woman’s body, fertilizing them with sperm in the laboratory and transferring the resulting embryos back in the woman’s body. |
| Azoospermia | Complete lack of sperm in the ejaculate. |
| Biallelic | Variants detected in both gene alleles located on homologous chromosomes. |
| Clinically actionable genetic variant | Likely pathogenic or pathogenic variants in genes associated with diseases that are moderately to highly penetrant. |
| ClinVar database | ClinVar is a public archive with free access to reports on the relationships between human genetic variations and phenotypes, with supporting evidence. |
| Congenital bilateral absence of the vas deferens (CBAVD) | Several parts of the reproductive tract (the vas deferens, most of the epididymis and seminal vesicles) are missing in both sides of the body from birth. |
| Congenital hypogonadotropic hypogonadism (CHH) | Congenital defects in the hypothalamic–pituitary-gonadal axis leading to impaired central hormonal regulation of testis function. Frequently correctable. |
| Cryptozoospermia | Spermatozoa detectable only after centrifugation of the semen sample. |
| Di- and oligogenic inheritance | Genetic disease caused by the combined effect of two (or a few) clinically actionable variants. |
| Diagnostic gene panel | List of genes analysed for clinically actionable genetic variants using sequencing or genotyping in patients presenting the relevant phenotype. |
| Diagnostic yield | The likelihood that a test will result in the diagnostic cause of the disease. |
| Disorders of sex development (DSD) | Any problem where the genitalia are atypical in relation to the gonads or chromosomes (46,XY DSD or 46,XX DSD). |
| Exome sequencing | Sequencing of the coding part of the genome (1–2%). |
| Globozoospermia | Sperm cells with round head and no acrosome. |
| Isolated infertility | Infertility without any other apparent health problems. |
| Likely pathogenic/pathogenic variant (LP/P) | There is a high likelihood (greater than 90% certainty) that a variant is disease-causing/Causative for a disease. |
| Loss-of-function variant (LoF) | Genetic variant leading to a truncated protein, including nonsense (STOP), frameshift (fs) and splicing variants. |
| Macrozoospermia | Large-headed multiflagellar spermatozoa. |
| Multiple morphological abnormalities of the sperm flagella (MMAF) | Sperm with a mosaic of various flagellar abnormalities, such as absent, short, bent, coiled, and irregular flagella. |
| Non-obstructive azoospermia (NOA) | Lack of sperm in the ejaculate caused by congenital or acquired testicular disorders or primary spermatogenic failure. Usually uncorrectable. |
| Aneuploidy | Abnormal number of chromosomes in a cell. |
| Aspermia | Complete lack of semen with ejaculation. |
| Assisted reproductive technology (ART) | Infertility treatment that involves removal of oocytes from a woman’s body, fertilizing them with sperm in the laboratory and transferring the resulting embryos back in the woman’s body. |
| Azoospermia | Complete lack of sperm in the ejaculate. |
| Biallelic | Variants detected in both gene alleles located on homologous chromosomes. |
| Clinically actionable genetic variant | Likely pathogenic or pathogenic variants in genes associated with diseases that are moderately to highly penetrant. |
| ClinVar database | ClinVar is a public archive with free access to reports on the relationships between human genetic variations and phenotypes, with supporting evidence. |
| Congenital bilateral absence of the vas deferens (CBAVD) | Several parts of the reproductive tract (the vas deferens, most of the epididymis and seminal vesicles) are missing in both sides of the body from birth. |
| Congenital hypogonadotropic hypogonadism (CHH) | Congenital defects in the hypothalamic–pituitary-gonadal axis leading to impaired central hormonal regulation of testis function. Frequently correctable. |
| Cryptozoospermia | Spermatozoa detectable only after centrifugation of the semen sample. |
| Di- and oligogenic inheritance | Genetic disease caused by the combined effect of two (or a few) clinically actionable variants. |
| Diagnostic gene panel | List of genes analysed for clinically actionable genetic variants using sequencing or genotyping in patients presenting the relevant phenotype. |
| Diagnostic yield | The likelihood that a test will result in the diagnostic cause of the disease. |
| Disorders of sex development (DSD) | Any problem where the genitalia are atypical in relation to the gonads or chromosomes (46,XY DSD or 46,XX DSD). |
| Exome sequencing | Sequencing of the coding part of the genome (1–2%). |
| Globozoospermia | Sperm cells with round head and no acrosome. |
| Isolated infertility | Infertility without any other apparent health problems. |
| Likely pathogenic/pathogenic variant (LP/P) | There is a high likelihood (greater than 90% certainty) that a variant is disease-causing/Causative for a disease. |
| Loss-of-function variant (LoF) | Genetic variant leading to a truncated protein, including nonsense (STOP), frameshift (fs) and splicing variants. |
| Macrozoospermia | Large-headed multiflagellar spermatozoa. |
| Multiple morphological abnormalities of the sperm flagella (MMAF) | Sperm with a mosaic of various flagellar abnormalities, such as absent, short, bent, coiled, and irregular flagella. |
| Non-obstructive azoospermia (NOA) | Lack of sperm in the ejaculate caused by congenital or acquired testicular disorders or primary spermatogenic failure. Usually uncorrectable. |
| Obstructive azoospermia (OA) | Congenital or acquired obstruction of vas deference, epididymis, or ejaculatory duct with normal spermatogenesis. Managed with assisted reproductive technologies. |
| Oligozoospermia | Impaired spermatogenesis; < 39 million sperm per ejaculate1. |
| Pleiotropy | One gene influences two or more seemingly unrelated phenotypic traits. |
| Penetrance | The proportion of people carrying a genetic variation, who exhibit signs and symptoms of a genetic disorder. |
| Recurrent microdeletion | Recurrent loss of the same chromosomal region in several patients. |
| Secondary findings | Genetic testing results that provide information about clinically actionable variants unrelated to the primary purpose of the testing. |
| Single nucleotide variant (SNV) | Substitution of a single DNA base pair. Mostly neutral changes. May be clinically actionable in case located in a coding or gene regulatory region. |
| Syndromic infertility | Infertility with other congenital health problems and comorbidities. |
| Testicular sperm extraction (TESE) | Surgical removing a small portion of testicular tissue and extracting any viable sperm for use in the ICSI procedure. |
| Obstructive azoospermia (OA) | Congenital or acquired obstruction of vas deference, epididymis, or ejaculatory duct with normal spermatogenesis. Managed with assisted reproductive technologies. |
| Oligozoospermia | Impaired spermatogenesis; < 39 million sperm per ejaculate1. |
| Pleiotropy | One gene influences two or more seemingly unrelated phenotypic traits. |
| Penetrance | The proportion of people carrying a genetic variation, who exhibit signs and symptoms of a genetic disorder. |
| Recurrent microdeletion | Recurrent loss of the same chromosomal region in several patients. |
| Secondary findings | Genetic testing results that provide information about clinically actionable variants unrelated to the primary purpose of the testing. |
| Single nucleotide variant (SNV) | Substitution of a single DNA base pair. Mostly neutral changes. May be clinically actionable in case located in a coding or gene regulatory region. |
| Syndromic infertility | Infertility with other congenital health problems and comorbidities. |
| Testicular sperm extraction (TESE) | Surgical removing a small portion of testicular tissue and extracting any viable sperm for use in the ICSI procedure. |
| Obstructive azoospermia (OA) | Congenital or acquired obstruction of vas deference, epididymis, or ejaculatory duct with normal spermatogenesis. Managed with assisted reproductive technologies. |
| Oligozoospermia | Impaired spermatogenesis; < 39 million sperm per ejaculate1. |
| Pleiotropy | One gene influences two or more seemingly unrelated phenotypic traits. |
| Penetrance | The proportion of people carrying a genetic variation, who exhibit signs and symptoms of a genetic disorder. |
| Recurrent microdeletion | Recurrent loss of the same chromosomal region in several patients. |
| Secondary findings | Genetic testing results that provide information about clinically actionable variants unrelated to the primary purpose of the testing. |
| Single nucleotide variant (SNV) | Substitution of a single DNA base pair. Mostly neutral changes. May be clinically actionable in case located in a coding or gene regulatory region. |
| Syndromic infertility | Infertility with other congenital health problems and comorbidities. |
| Testicular sperm extraction (TESE) | Surgical removing a small portion of testicular tissue and extracting any viable sperm for use in the ICSI procedure. |
| Obstructive azoospermia (OA) | Congenital or acquired obstruction of vas deference, epididymis, or ejaculatory duct with normal spermatogenesis. Managed with assisted reproductive technologies. |
| Oligozoospermia | Impaired spermatogenesis; < 39 million sperm per ejaculate1. |
| Pleiotropy | One gene influences two or more seemingly unrelated phenotypic traits. |
| Penetrance | The proportion of people carrying a genetic variation, who exhibit signs and symptoms of a genetic disorder. |
| Recurrent microdeletion | Recurrent loss of the same chromosomal region in several patients. |
| Secondary findings | Genetic testing results that provide information about clinically actionable variants unrelated to the primary purpose of the testing. |
| Single nucleotide variant (SNV) | Substitution of a single DNA base pair. Mostly neutral changes. May be clinically actionable in case located in a coding or gene regulatory region. |
| Syndromic infertility | Infertility with other congenital health problems and comorbidities. |
| Testicular sperm extraction (TESE) | Surgical removing a small portion of testicular tissue and extracting any viable sperm for use in the ICSI procedure. |
Conclusions
Screening for karyotype abnormalities, pathogenic variants in the CFTR gene and Y-chromosomal microdeletions has been routine practice in andrology for >20 years, explaining up to 10% of infertility cases.
Rare specific forms of male infertility, such as CHH, CBAVD, DSD and qualitative sperm defects, are caused by recurrently affected genes, which facilitate molecular diagnostics based on targeted gene panels.
Monogenic causes of spermatogenic failure and impairment are heterogeneous, thus ES is currently the most optimal approach to detect the genetic cause in each clinical case.
All clinical cases along with the identified genetic variants need to be reported in scientific literature and medical genetics databases (e.g. ClinVar, OMIM) to increase the confidence in diagnostics.
Clinical guidelines developed by international teams with multidisciplinary expertise are needed for smooth integration of expanded molecular diagnostics in the routine management of infertile men
More basic research is needed to uncover the role of di−/oligogenic inheritance, structural variants and common genetic variation in modulating male fertility potential.
Uncovering novel genes and biological pathways implicated in spermatogenic failure may lead to innovative and preferentially non-invasive treatment targets and management measures for male infertility.
Data availability statement
No new data were generated or analysed in support of this review.
Funding
Preparation of the review was supported by the Estonian Research Council grant PRG1021.
References
Houston BJ, Riera-Escamilla A, Wyrwoll MJ, et al.

{kind=link}
{kind=link}