





Summary
Rare variants in the genes IL36RN, CARD14 and AP1S3 have been identified to cause or contribute to pustular skin diseases, primarily generalized pustular psoriasis (GPP).
To better understand the disease relevance of these genes, we screened our cohorts of patients with pustular skin diseases [primarily GPP and palmoplantar pustular psoriasis (PPP)] for coding changes in these three genes. Carriers of single heterozygous IL36RN mutations were screened for a second mutation in IL36RN.
Coding exons of IL36RN, CARD14 and AP1S3 were sequenced in 67 patients – 61 with GPP, two with acute generalized exanthematous pustulosis and four with acrodermatitis continua of Hallopeau. We screened IL36RN and AP1S3 for intragenic copy‐number variants and 258 patients with PPP for coding changes in AP1S3. Eleven heterozygous IL36RN mutations carriers were analysed for a second noncoding IL36RN mutation. Genotype–phenotype correlations in carriers/noncarriers of IL36RN mutations were assessed within the GPP cohort.
The majority of patients (GPP, 64%) did not carry rare variants in any of the three genes. Biallelic and monoallelic IL36RN mutations were identified in 15 and five patients with GPP, respectively. Noncoding rare IL36RN variants were not identified in heterozygous carriers. The only significant genotype–phenotype correlation observed for IL36RN mutation carriers was early age at disease onset. Additional rare CARD14 or AP1S3 variants were identified in 15% of IL36RN mutation carriers.
The identification of IL36RN mutation carriers harbouring additional rare variants in CARD14 or AP1S3 indicates a more complex mode of inheritance of pustular psoriasis. Our results suggest that, in heterozygous IL36RN mutation carriers, there are additional disease‐causing genetic factors outside IL36RN.
Within the past few years, variants in three genes have been identified to be involved in the pathogenesis of pustular psoriasis and other pustular skin diseases. Biallelic mutations in IL36RN have been described as disease‐causing in 21–41% of Asian/European patients with generalized pustular psoriasis (GPP),1,2,3 indicating IL36RN mutations as a major pathogenic factor in GPP.4,5 In some patients only a single mutation can be identified. Mutations in IL36RN lead to a reduced function or loss of function of the interleukin (IL)‐36 receptor‐antagonist (IL‐36Ra) and therefore to an imbalance in the IL‐36 pathway in favour of proinflammatory IL‐36 cytokines. This imbalance results in increased activation of the IL‐36 pathway with subsequent activation of the mitogen‐activated protein kinases and production of, for example, IL‐8 and IL‐6.4 The same IL36RN mutations are also associated with other pustular skin diseases, acrodermatitis continua of Hallopeau (ACH) and acute generalized exanthematous pustulosis (AGEP),6,7 predominantly as single heterozygous mutations. Evidence for an association with palmoplantar pustular psoriasis (PPP) is less consistent8,9 and no association was identified with more common forms of psoriasis.10,11
Genetic variants in CARD14 have also been implicated as disease‐causing/disease‐contributing factors in patients with GPP and PPP;12,13,14,15,in vitro experiments of most of these variants suggest an activation of nuclear factor kappa B (NF‐κB).12,13 Two missense variants in AP1S3 have further been described to contribute to ACH, GPP and PPP6,16 and in vitro analysis suggests a reduced protein function resulting in disruption of keratinocyte autophagy and an increased activation of NF‐κB leading to upregulation of IL‐1 signalling and overexpression of IL‐36α.6,16
Because for IL36RN and AP1S3 genetic variants have been shown to lead to a reduced function/loss of function of the corresponding protein, copy‐number variants (CNVs) affecting coding parts of genes were considered as candidate variants. However, screening for CNVs has rarely been included in genetic studies of pustular skin diseases.8,14
To better understand the disease relevance of the three genes, we screened our patient cohorts with pustular skin diseases for qualitative coding variants in the three genes and copy‐number variants of coding exons in IL36RN and AP1S3. In addition, we performed genotype–phenotype correlations in carriers/noncarriers of IL36RN mutations within the GPP cohort. Finally, heterozygous carriers of a single IL36RN mutation were analysed for noncoding rare variants in IL36RN as candidates for a second disease‐causing variant.
Patients and methods
Patients
We screened our patient cohorts with pustular skin diseases for coding variants in three genes. These cohorts comprised 61 patients with GPP, two with AGEP and four with ACH, as well as 258 patients with PPP. Individual characteristics of the participants with GPP, AGEP and ACH patients are listed in Table S1 (see Supporting Information). The patient groups were enlarged compared with previous studies,8,14 including 55 additional patients (n =42 for GPP, n =2 for AGEP, n =4 in ACH, n =7 for PPP).
All patients’ diagnoses were made by certified dermatologists according to current knowledge of diseases; the majority of patients with pustular psoriasis had been known to the recruiting centres over longer periods of time before recruitment. Patients with pustular skin disease as a possible paradoxical reaction to a biological treatment were excluded from participation in this study. The diagnosis of PPP was made on the basis of the typical clinical picture, and only patients with unequivocal PPP were included in the study. The diagnosis of ACH was also made clinically by means of a disease course supporting the diagnosis (e.g. chronic disease course, negative bacterial swabs or lack of response to antibiotic therapy and treatment response to immunosuppressive drugs). The diagnosis of GPP was based on the clinical picture, which often includes symptoms of concomitant systemic inflammation and hypoparathyroidism or hypocalcaemia, and was chronic or recurrent in all but four out of 61 cases (Table S1). AGEP was also diagnosed clinically and made in cases of patients with a single episode of a pustular rash in the presence of a suspicious drug or the absence of a personal or family history of psoriasis.
Most of the patients with GPP were of European origin (n =51), while in sum, 10 patients were from other regions of the world (four patients from Turkey, one from Syria, one from Morocco, one from Palestine, one from a foreign country not further specified, and two patients had grandparents from Iraq) (Table S1). All patients with AGEP and ACH except patient AGEP01 (Siberia) were of European origin (Table S1). Within the group of 258 patients with PPP, four patients were not of European origin (two from Turkey, one from Africa and one from a foreign country not further specified). As previously reported,8 of the 254 European patients in the PPP group there were 34 Estonians.
Molecular analyses
Patients with GPP, AGEP and ACH were sequenced for all coding exons of IL36RN and CARD14 as described previously,14 and we included previously published genetic data on subgroups of our cohorts in the present analyses. In addition, we analysed the 325 patients (including the PPP group) for all coding exons of AP1S3 by Sanger sequencing. The seven newly included patients with PPP were screened for coding variation in IL36RN and CARD14. Furthermore, we sequenced 11 heterozygous carriers of IL36RN mutations for noncoding exons, intronic sequences, the 3ʹ untranslated region (UTR, in sum ~5·8 kilobases) as well as upstream of the gene (2·45 kilobases) by Sanger sequencing using 26 overlapping polymerase chain reaction products.
In addition, a quantitative analysis of all coding parts of IL36RN was performed on all patients’ DNA as described previously,14 and we established a new quantitative multiplex ligation‐dependent probe amplification test for all coding exons of AP1S3 according to the recommendations of the manufacturer (MRC‐Holland, Amsterdam, the Netherlands).17 Detailed phenotypic data from the patients with GPP, AGEP and ACH allowed for genotype–phenotype correlations (Table S1).
Comparison with controls and statistical analyses
We analysed frequencies of all identified variants in the currently largest possible control cohort of European individuals and excluded variants with a minor allele frequency of > 2%.
As a result of low frequencies of coding variants, Fisher's exact test was applied to determine allele frequency differences between exclusively European individuals and controls. Fisher's exact test was also applied to determine differences between carriers of IL36RN mutations and wild‐type carriers. The Wilcoxon rank test was used for comparison of age at onset between those carriers and noncarriers of IL36RN mutations. All statistical analyses were performed with the software R (http://www.r-project.org/).18
Molecular modelling
Molecular modelling of variants in IL36RN was investigated on the basis of a high‐resolution crystal structure of a loop‐swapped IL‐36Ra (PDB: 4P0L).19 The swapped loops are located distally from the sites of mutations investigated in the present study, and therefore the respective structure was considered as a suitable model for wild‐type IL‐36Ra. The p.Val44Met exchange was modelled with Swiss‐PDB‐Viewer (https://spdbv.vital-it.ch/)20 and the lowest‐energy rotamer was selected for the Met44 side chain. The Thr99/Phe100 deletion was modelled with ModLoop (https://modbase.compbio.ucsf.edu/modloop/).21,22 In the case of the p.Thr22Ala variant in AP1S3, its effect was modelled using Swiss‐PDB‐Viewer based on the known crystal structure (PDB: 4HMY).23 RasMol (http://www.openrasmol.org/ )24 was used for structure analysis and visualization of both proteins.
Further assessments of rare variants
Rare variants were classified as disease‐relevant or ‐irrelevant according to previously published experimental data and/or to results of molecular modelling as described previously14 including previously unreported variants (see above). In addition, to assess all missense variants in IL36RN further, we used dbNSFP v2·0 (https://sites.google.com/site/jpopgen/dbNSFP)25 resulting in a combination of different protein prediction tools. In case of the inframe deletion c.295‐300delACCTTC/p.Thr99_Phe100del, the combined annotation dependent depletion (CADD) score was generated with the CADD tool (http://cadd.gs.washington.edu/info).26 Furthermore, we interpreted all missense variants identified and the inframe deletion in IL36RN according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines with the Genetic Variant Interpretation Tool (http://www.medschool.umaryland.edu/Genetic_Variant_Interpretation_Tool1.html/).27 Those criteria included, for example, the previous study by Tauber et al.28 providing substantial evidence for functional effects in in vitro analyses, their rarity in large exome sequencing projects (Table S2; see Supporting Information) as well as their detection in trans with a pathogenic variant.
Results
Generalized pustular psoriasis
The majority (n =43, 71%) of patients with GPP were female; five patients had received the diagnosis of GPP as one of several differential diagnoses (Table S1). The mean age at recruitment was 47·5 ± 17·5 years, the mean age of disease onset 33·4 ± 22·4 years. The majority of patients did not carry a rare nonsynonymous variant in any of the three investigated genes (64%, Fig. 1a), but rare nonsynonymous IL36RN variants were identified in 20 patients with GPP (33%), biallelic IL36RN mutations in 15 patients with GPP (25%) and single mutations in five individuals (8%) (Table S1). A total of 11 different mutations were detected (Fig. 1b). The most frequent mutation was c.333>C>T (p.Ser113Leu) followed by c.227C>T (p.Pro76Leu). We identified four variants in IL36RN so far unreported in pustular psoriasis: c.338C>A (p.Ser113X), c.295‐300delACCTTC (p.Thr99_Phe100del), c.130G>A (p.Val44Met) and c.308G>A (p.Arg103Gln) (Table S1). According to molecular modelling, c.338C>A (p.Ser113X) resulted in a shortened, destabilized protein analogous to c.280G>T (p.Glu94X).14Val44 is located in the hydrophobic core of IL‐36Ra (Fig. 2a).In the Val44Met variant, the longer Met44 side chain causes steric clashes with Phe151 (Fig. 2b), which are expected to decrease protein stability. Arg103 is solvent‐exposed in the IL‐36RA structure and does not form any specific interactions with the adjacent side chains. Thus, mutation of Arg103 appears less critical than that of the adjacent R102, which forms numerous stabilizing interactions. This is in line with the sequence‐based classification that predicts the Arg102Trp more critical than the Arg103Gln exchange.
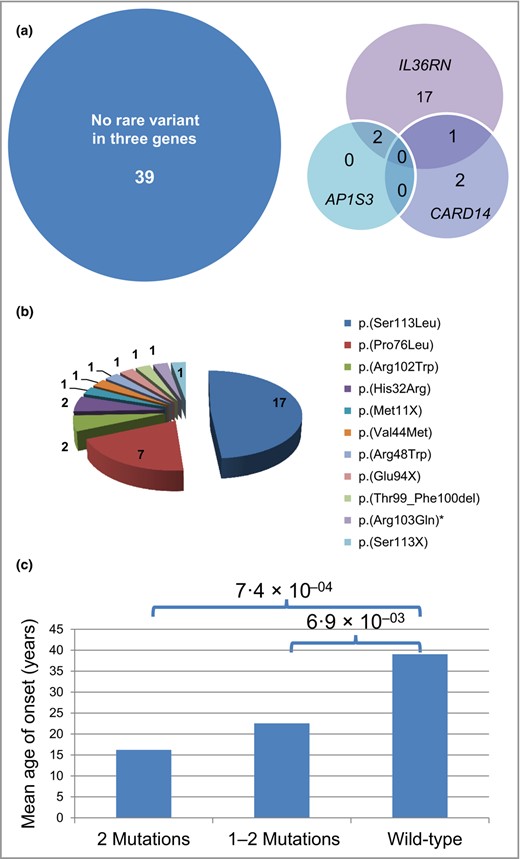
Mutations in IL36RN and clinical correlations in 61 patients with generalized pustular psoriasis (GPP). (a) Venn diagram indicating overall number of carriers of mutations in IL36RN, of rare variants in AP1S3 or CARD14, of carriers of rare variants in two of these genes and of noncarriers of a variant in any of the three genes in patients with GPP. (b) Distribution of IL36RN mutations in 61 patients with GPP. * indicates the only rare variant classified as a variant of unknown significance. (c) Correlation of mutation status in IL36RN (x‐axis) with age at manifestation (y‐axis). ‘2 Mutations’ indicates carriers of mutations on both gene copies, ‘1–2 Mutations’ carriers of a single mutation as well as carriers of mutations on both gene copies, and ‘wild‐type’ carriers are those with no IL36RN mutations.
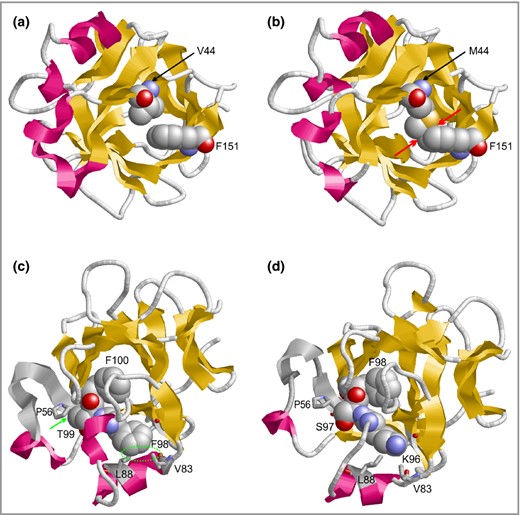
Molecular modelling of novel variants in IL36Ra. Effect of the Val44Met exchange and the Thr99/Phe100 deletion on the structure of IL36Ra. (a) Val44 is located in the hydrophobic core of the molecule. (b) Met44 forms clashes with Phe151 that are not present in the wild‐type. The side chains of residues 44 and 151 are shown in space‐filling presentation and steric clashes are indicated by red arrows. (c) Location and interaction of residues Phe98, Thr99 and Phe100 (all shown in space‐filling presentation) in the wild‐type. Hydrophobic interactions between Thr99 and Pro56 as well as Phe98 and Val83/Leu88 are denoted with a green arrow and circle, respectively. (d) Thr99/Phe100 mutant, in which Phe98 occupies the position of Phe100 from the wild‐type. Note the loss of hydrophobic interactions of residues Lys96/Ser97 compared with Phe98/Thr99 in the wild‐type.
Thr99 and Phe100 are rather buried in the IL‐36Ra structure and form tight interactions with adjacent residues (Fig. 2c). Modelling suggests that upon deletion of Thr99/Phe100 the respective spatial positions become occupied by the N‐terminally adjacent residues Ser97/Phe98, which would allow Phe98 to establish the same crucial interactions observed for Phe100 in the wild‐type (Fig. 2c, d). Ser97 lacks the side‐chain methyl group present in Thr99, which results in a loss of hydrophobic interactions with Pro56 (Fig. 2c, d). However, the strongest loss of stability is observed around the site at which Phe98 is located in the wild‐type. Phe98 forms tight hydrophobic contacts with Val83 and Leu88 in the wild‐type protein thereby stabilizing the position of the respective helix (Fig. 2c). In the deletion mutant, Lys96 is shifted to the respective position of the IL‐36Ra structure (Fig. 2d), which causes a loss of the hydrophobic contacts and which is expected to cause a decrease in protein stability. Therefore, c.130G>A (p.Val44Met) and c.295‐300delACCTTC (p.Thr99_Phe100del) were also predicted to result in destabilized, likely disease‐relevant, loss‐of‐function proteins (Fig. 2).
When we applied further assessments of those rare genetic variants using protein prediction programs, we observed fewer harmful predictions in the case of c.130G>A (p.Val44Met) compared with other known disease‐causing variants (Table S3; see Supporting Information). Interestingly, the same analyses for some of the established harmful rare missense variants in IL36RN such as c.333>C>T (p.Ser113Leu) – shown to result in reduced protein levels in in vitro analyses28 – did not provide evidence for a harmful effect with the majority of algorithms. Furthermore, as pathogenicity of rare coding variants in a monogenic disease cannot be predicted by using those algorithms alone, we assessed the pathogenicity of so far unreported rare variants – including the inframe deletion – by using ACMG standards and guidelines. Thereby, the variants c.130G>A (p.Val44Met) and c.295‐300delACCTTC (p.Thr99_Phe100del) were classified as likely pathogenic (class IV variants) and c.308G>A (p.Arg103Gln) was classified as a variant of unknown significance. As this latter classification neither excludes nor proves a disease‐causing status and the overall proportion of this variant in the sum of mutated IL36RN alleles was minor, we stayed with this variant for further analyses.
Heterozygous AP1S3 mutations were detected in two patients with GPP, both of whom carried additional homozygous or compound‐heterozygous IL36RN mutations (Fig. 1a, Table 1, Table S1). A daughter of a consanguineous marriage carried a homozygous IL36RN mutation as well as the homozygous AP1S3 variant c.64A>G (p.Thr22Ala) [minor allele frequency in Exome Aggregation Consortium (120 618 alleles): 0·17%]. According to molecular modelling, the Ala22 mutant protein loses two intermolecular contacts, weakening the interaction between the γ‐1 chain and the σ‐3 chain (Fig. S1). Although potentially relevant in a homozygous state, the variant was excluded from statistical analyses, as it is probably less functionally relevant than the c.11T>G (p.Phe4Cys) and c.97C>T (p.Arg33Trp) variants identified previously.6 Furthermore, none of the used protein prediction tools considered this effect as harmful, confirming a previous analysis6 and strengthening our strategy of exclusion. Heterozygous rare missense variants in CARD14 were identified in four patients with GPP; but we excluded c.599G>A (p.Ser200Asn) from further analysis, as it has been described to be a missense variant without significantly differential effects on activation of NF‐κB in comparison with wild‐type.29 One of the further three missense variants has been previously associated with GPP and PPP.8,12,15 Of note, one of the three patients (GPP18) carried an additional mutation in IL36RN (Fig. 1a, Table S1).
Association analysis of different pustular skin diseases with rare variants in AP1S3, CARD14 and IL36RN in European individualsa
| Gene, variant | GPP (n =51) | AGEP (n =1) | ACH (n =4) | PPP (n =254) | ∑ Pustular psoriasis phenotypes (n =310) | Control groupb |
| AP1S3 | ||||||
| c.11T>G (p.Phe4Cys) | 1 (1·0) | 0 (0) | 0 (0) | 10 (2·0) | 11 (1·8) | 778 (1·2) |
| c.97C>T (p.Arg33Trp) | 1 (1·0) | 1 (0·5) | 0 (0) | 3 (0·6) | 5 (0·8) | 724 (1·1) |
| ∑ mutant alleles | 3 (2·7) | 13 (2·6) | 16 (2·6) | 1,502 (2·3) | ||
| ∑ wild‐type alleles | 109 (97·3) | 495 (97·4) | 604 (97·4) | 64 226 (97·7) | ||
| P‐value | 0·74 | 0·65 | 0·59 | |||
| CARD14 | ||||||
| c.206G>A (p.Arg69Gln) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 1 (0·007) |
| c.349G>A (p.Gly117Ser) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 0 (0) |
| c.526G>C (p.Asp176His) | 0 (0) | 0 (0) | 0 (0) | 2 (0·4) | 2 (0·31) | 7 (0·05) |
| c.536G>A (p.Arg179His) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 3 (0·02) |
| ∑ mutant alleles | 3 (2·7) | 2 (0·4) | 5 (0·8) | 11 (0·08) | ||
| ∑ wild‐type alleles | 109 (97·3) | 506 (99·6) | 615 (99·2) | 13 519 (99·92) | ||
| P‐value | 1·8 × 10−04 | 0·08 | 4·6 × 10−04 | |||
| IL36RN | ||||||
| Mutant alleles | 25 (24·5) | 0 (0) | 2 (0·25) | 3 (0·6) | 30 (4·8) | 358 (0·54) |
| ∑ mutant alleles | 27 (24·1) | 3 (0·6) | 30 (4·8) | 358 (0·54) | ||
| ∑ wild‐type alleles | 85 (75·9) | 505 (99·4) | 590 (95·2) | 65 384 (99·46) | ||
| P‐value | < 2·2 × 10−16 | 0·76 | < 2·2 × 10−16 | |||
| Gene, variant | GPP (n =51) | AGEP (n =1) | ACH (n =4) | PPP (n =254) | ∑ Pustular psoriasis phenotypes (n =310) | Control groupb |
| AP1S3 | ||||||
| c.11T>G (p.Phe4Cys) | 1 (1·0) | 0 (0) | 0 (0) | 10 (2·0) | 11 (1·8) | 778 (1·2) |
| c.97C>T (p.Arg33Trp) | 1 (1·0) | 1 (0·5) | 0 (0) | 3 (0·6) | 5 (0·8) | 724 (1·1) |
| ∑ mutant alleles | 3 (2·7) | 13 (2·6) | 16 (2·6) | 1,502 (2·3) | ||
| ∑ wild‐type alleles | 109 (97·3) | 495 (97·4) | 604 (97·4) | 64 226 (97·7) | ||
| P‐value | 0·74 | 0·65 | 0·59 | |||
| CARD14 | ||||||
| c.206G>A (p.Arg69Gln) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 1 (0·007) |
| c.349G>A (p.Gly117Ser) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 0 (0) |
| c.526G>C (p.Asp176His) | 0 (0) | 0 (0) | 0 (0) | 2 (0·4) | 2 (0·31) | 7 (0·05) |
| c.536G>A (p.Arg179His) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 3 (0·02) |
| ∑ mutant alleles | 3 (2·7) | 2 (0·4) | 5 (0·8) | 11 (0·08) | ||
| ∑ wild‐type alleles | 109 (97·3) | 506 (99·6) | 615 (99·2) | 13 519 (99·92) | ||
| P‐value | 1·8 × 10−04 | 0·08 | 4·6 × 10−04 | |||
| IL36RN | ||||||
| Mutant alleles | 25 (24·5) | 0 (0) | 2 (0·25) | 3 (0·6) | 30 (4·8) | 358 (0·54) |
| ∑ mutant alleles | 27 (24·1) | 3 (0·6) | 30 (4·8) | 358 (0·54) | ||
| ∑ wild‐type alleles | 85 (75·9) | 505 (99·4) | 590 (95·2) | 65 384 (99·46) | ||
| P‐value | < 2·2 × 10−16 | 0·76 | < 2·2 × 10−16 | |||
Numbers indicate absolute allele counts (percentages). w/o, without. aMutant and wild‐type alleles of variants in 51 patients with generalized pustular psoriasis (GPP), one with acute generalized exanthematous pustulosis (AGEP) patient, four with acrodermatitis continua of Hallopeau (ACH), 254 with palmoplantar pustular psoriasis (PPP) and (database) controls as well as results of association analysis are shown. b32 864 Non‐Finnish European individuals in the publicly available database, Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/),30 with available genotypes of the two AP1S3 variants, including 12 homozygous carriers of c.11T>G (p.Phe4Cys) and three homozygous carriers of c.97C>T (p.Arg33Trp) and of the IL36RN mutations (see also Table S2; see Supporting Information). Coverage of CARD14 in ExAC was not as good as in AP1S3 and IL36RN, therefore a combination of different control groups was used, similarly as described in Mossner et al.8 and indicated in detail in Table S5 (see Supporting Information).
Association analysis of different pustular skin diseases with rare variants in AP1S3, CARD14 and IL36RN in European individualsa
| Gene, variant | GPP (n =51) | AGEP (n =1) | ACH (n =4) | PPP (n =254) | ∑ Pustular psoriasis phenotypes (n =310) | Control groupb |
| AP1S3 | ||||||
| c.11T>G (p.Phe4Cys) | 1 (1·0) | 0 (0) | 0 (0) | 10 (2·0) | 11 (1·8) | 778 (1·2) |
| c.97C>T (p.Arg33Trp) | 1 (1·0) | 1 (0·5) | 0 (0) | 3 (0·6) | 5 (0·8) | 724 (1·1) |
| ∑ mutant alleles | 3 (2·7) | 13 (2·6) | 16 (2·6) | 1,502 (2·3) | ||
| ∑ wild‐type alleles | 109 (97·3) | 495 (97·4) | 604 (97·4) | 64 226 (97·7) | ||
| P‐value | 0·74 | 0·65 | 0·59 | |||
| CARD14 | ||||||
| c.206G>A (p.Arg69Gln) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 1 (0·007) |
| c.349G>A (p.Gly117Ser) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 0 (0) |
| c.526G>C (p.Asp176His) | 0 (0) | 0 (0) | 0 (0) | 2 (0·4) | 2 (0·31) | 7 (0·05) |
| c.536G>A (p.Arg179His) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 3 (0·02) |
| ∑ mutant alleles | 3 (2·7) | 2 (0·4) | 5 (0·8) | 11 (0·08) | ||
| ∑ wild‐type alleles | 109 (97·3) | 506 (99·6) | 615 (99·2) | 13 519 (99·92) | ||
| P‐value | 1·8 × 10−04 | 0·08 | 4·6 × 10−04 | |||
| IL36RN | ||||||
| Mutant alleles | 25 (24·5) | 0 (0) | 2 (0·25) | 3 (0·6) | 30 (4·8) | 358 (0·54) |
| ∑ mutant alleles | 27 (24·1) | 3 (0·6) | 30 (4·8) | 358 (0·54) | ||
| ∑ wild‐type alleles | 85 (75·9) | 505 (99·4) | 590 (95·2) | 65 384 (99·46) | ||
| P‐value | < 2·2 × 10−16 | 0·76 | < 2·2 × 10−16 | |||
| Gene, variant | GPP (n =51) | AGEP (n =1) | ACH (n =4) | PPP (n =254) | ∑ Pustular psoriasis phenotypes (n =310) | Control groupb |
| AP1S3 | ||||||
| c.11T>G (p.Phe4Cys) | 1 (1·0) | 0 (0) | 0 (0) | 10 (2·0) | 11 (1·8) | 778 (1·2) |
| c.97C>T (p.Arg33Trp) | 1 (1·0) | 1 (0·5) | 0 (0) | 3 (0·6) | 5 (0·8) | 724 (1·1) |
| ∑ mutant alleles | 3 (2·7) | 13 (2·6) | 16 (2·6) | 1,502 (2·3) | ||
| ∑ wild‐type alleles | 109 (97·3) | 495 (97·4) | 604 (97·4) | 64 226 (97·7) | ||
| P‐value | 0·74 | 0·65 | 0·59 | |||
| CARD14 | ||||||
| c.206G>A (p.Arg69Gln) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 1 (0·007) |
| c.349G>A (p.Gly117Ser) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 0 (0) |
| c.526G>C (p.Asp176His) | 0 (0) | 0 (0) | 0 (0) | 2 (0·4) | 2 (0·31) | 7 (0·05) |
| c.536G>A (p.Arg179His) | 1 (1·0) | 0 (0) | 0 (0) | 0 (0) | 1 (0·15) | 3 (0·02) |
| ∑ mutant alleles | 3 (2·7) | 2 (0·4) | 5 (0·8) | 11 (0·08) | ||
| ∑ wild‐type alleles | 109 (97·3) | 506 (99·6) | 615 (99·2) | 13 519 (99·92) | ||
| P‐value | 1·8 × 10−04 | 0·08 | 4·6 × 10−04 | |||
| IL36RN | ||||||
| Mutant alleles | 25 (24·5) | 0 (0) | 2 (0·25) | 3 (0·6) | 30 (4·8) | 358 (0·54) |
| ∑ mutant alleles | 27 (24·1) | 3 (0·6) | 30 (4·8) | 358 (0·54) | ||
| ∑ wild‐type alleles | 85 (75·9) | 505 (99·4) | 590 (95·2) | 65 384 (99·46) | ||
| P‐value | < 2·2 × 10−16 | 0·76 | < 2·2 × 10−16 | |||
Numbers indicate absolute allele counts (percentages). w/o, without. aMutant and wild‐type alleles of variants in 51 patients with generalized pustular psoriasis (GPP), one with acute generalized exanthematous pustulosis (AGEP) patient, four with acrodermatitis continua of Hallopeau (ACH), 254 with palmoplantar pustular psoriasis (PPP) and (database) controls as well as results of association analysis are shown. b32 864 Non‐Finnish European individuals in the publicly available database, Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/),30 with available genotypes of the two AP1S3 variants, including 12 homozygous carriers of c.11T>G (p.Phe4Cys) and three homozygous carriers of c.97C>T (p.Arg33Trp) and of the IL36RN mutations (see also Table S2; see Supporting Information). Coverage of CARD14 in ExAC was not as good as in AP1S3 and IL36RN, therefore a combination of different control groups was used, similarly as described in Mossner et al.8 and indicated in detail in Table S5 (see Supporting Information).
Other pustular skin diseases
For AGEP, both patients carried a heterozygous variant in AP1S3; in ACH, two of the four patients carried a heterozygous mutation in IL36RN (Table S1). Clinical characteristics of the patients with PPP (Table S4) were very similar to the ones reported in a largely overlapping group in PPP; 13 of 258 patients were heterozygous carriers harbouring one of the two nonsynonymous changes in AP1S3 (Table 1). Variant c.95C>T (p.Thr32Ile) was identified in two patients in the PPP group, but not included in further analyses, as molecular modelling did not predict a clear functional impact.
Association and quantitative analyses
Overall, we observed a highly significant association of pustular skin disease with IL36RN variants (P < 2·2 × 10−16) and a significant association of GPP with CARD14 variants (3·1 × 10−04) (Table 1). In contrast, we did not find any evidence of an association between AP1S3 variants and GPP, ACH or PPP (Table 1), and the groups of patients with AGEP and ACH on their own (n =2 and 4, respectively) are too small to draw any conclusions. By excluding c.599G>A (p.Ser200Asn) as a probably nonfunctional variant, association of PPP with coding variants in CARD14 was no longer identified.8 None of our patients was found to carry an intragenic deletion or duplication in IL36RN (n =325) or the coding regions of AP1S3 (n =322), with quantitative analysis of AP1S3 not resulting in reliable copy number results in three patients with PPP.
Analysis of noncoding regions of IL36RN
By sequencing the noncoding parts of IL36RN in all carriers of a single heterozygous mutation (n =11) identified in the current study or in a previous analysis of patients with PPP,8 we did not detect a single rare variant. These results in combination with the lack of disease‐causing intragenic CNVs render a disease‐relevant variant on the second IL36RN allele unlikely.
Genotype–phenotype correlation in generalized pustular psoriasis
Our genotype–phenotype correlation revealed a similar sex distribution in carriers of IL36RN mutations and wild‐type carriers, but a strong association between biallelic mutations in IL36RN and early age of manifestation (P =7·4 × 10−04). Also, the group of both carriers of single IL36RN mutations as well as of carriers of compound‐heterozygous/homozygous mutations had a significantly younger age at onset vs. noncarriers (P =6·9 × 10−03, Fig. 1c). We did not observe a significant difference in the frequency of accompanying psoriasis vulgaris in carriers/noncarriers of IL36RN mutations (Table 2). As in other autosomal recessively inherited mutations, the frequency of parental consanguinity was significantly higher in patients with two IL36RN mutations compared with noncarriers (Table 2). Furthermore, in patients with GPP, we observed no difference in carriers/noncarriers of IL36RN mutations with respect to frequency of episodic vs. continuous course of disease, positive family history for either psoriatic or rheumatic disease or frequency of other accompanying manifestations such as PPP, nail, joint or tongue involvement (Table 2).
Correlation analysis of carriers of two IL36RN mutations or one to two IL36RN mutations vs. noncarriers in 61 patients with generalized pustular psoriasis (GPP)
| Category and subcategory | Group A, carriers of two IL36RN mutations (n =15) | Group B, carriers of one to two IL36RN mutations (n =20) | Group C, noncarriers of IL36RN mutation (n =41) | P‐value (A vs. C) | P‐value (B vs. C) |
| Sex | |||||
| Male | 4 (27) | 5 (25) | 13 (32) | 1 | 0·76 |
| Female | 11 (73) | 15 (75) | 28 (68) | ||
| Course of diseasea | |||||
| Episodic | 13 (93) | 15 (83) | 26 (70) | 0·14 | 0·34 |
| Continuous | 1 (7) | 3 (17) | 11 (30) | ||
| Palmoplantar pustular psoriasis | |||||
| Yes | 3 (20) | 5 (25) | 4 (10) | 0·36 | 0·13 |
| No | 12 (80) | 15 (75) | 37 (90) | ||
| Tongue affectedb | |||||
| Yes | 4 (27) | 5 (25) | 8 (21) | 0·71 | 0·74 |
| No | 11 (73) | 15 (75) | 31 (79) | ||
| Psoriasis vulgarisc | |||||
| Yes | 3 (20) | 5 (25) | 20 (50) | 0·066 | 0·095 |
| No | 12 (80) | 15 (75) | 20 (50) | ||
| Nails affected | |||||
| Yes | 5 (33) | 5 (25) | 11 (27) | 0·74 | 1 |
| No | 10 (67) | 15 (75) | 30 (73) | ||
| Joints affected | |||||
| Yes | 4 (27) | 6 (30) | 12 (29) | 1 | 1 |
| No | 11 (73) | 14 (70) | 29 (71) | ||
| Cholangitis | |||||
| Yes | 1 (7) | 1 (5) | 1 (2) | 0·46 | 1 |
| No | 14 (93) | 19 (95) | 40 (98) | ||
| Family history positived | |||||
| Yes | 9 (60) | 11 (58) | 17 (43) | 0·36 | 0·4 |
| No | 6 (40) | 8 (42) | 23 (58) | ||
| Consanguinity | |||||
| Yes | 3 (20) | 3 (15) | 0 (0) | 0·016 | 0·032 |
| No | 12 (80) | 17 (85) | 41 (100) | ||
| Category and subcategory | Group A, carriers of two IL36RN mutations (n =15) | Group B, carriers of one to two IL36RN mutations (n =20) | Group C, noncarriers of IL36RN mutation (n =41) | P‐value (A vs. C) | P‐value (B vs. C) |
| Sex | |||||
| Male | 4 (27) | 5 (25) | 13 (32) | 1 | 0·76 |
| Female | 11 (73) | 15 (75) | 28 (68) | ||
| Course of diseasea | |||||
| Episodic | 13 (93) | 15 (83) | 26 (70) | 0·14 | 0·34 |
| Continuous | 1 (7) | 3 (17) | 11 (30) | ||
| Palmoplantar pustular psoriasis | |||||
| Yes | 3 (20) | 5 (25) | 4 (10) | 0·36 | 0·13 |
| No | 12 (80) | 15 (75) | 37 (90) | ||
| Tongue affectedb | |||||
| Yes | 4 (27) | 5 (25) | 8 (21) | 0·71 | 0·74 |
| No | 11 (73) | 15 (75) | 31 (79) | ||
| Psoriasis vulgarisc | |||||
| Yes | 3 (20) | 5 (25) | 20 (50) | 0·066 | 0·095 |
| No | 12 (80) | 15 (75) | 20 (50) | ||
| Nails affected | |||||
| Yes | 5 (33) | 5 (25) | 11 (27) | 0·74 | 1 |
| No | 10 (67) | 15 (75) | 30 (73) | ||
| Joints affected | |||||
| Yes | 4 (27) | 6 (30) | 12 (29) | 1 | 1 |
| No | 11 (73) | 14 (70) | 29 (71) | ||
| Cholangitis | |||||
| Yes | 1 (7) | 1 (5) | 1 (2) | 0·46 | 1 |
| No | 14 (93) | 19 (95) | 40 (98) | ||
| Family history positived | |||||
| Yes | 9 (60) | 11 (58) | 17 (43) | 0·36 | 0·4 |
| No | 6 (40) | 8 (42) | 23 (58) | ||
| Consanguinity | |||||
| Yes | 3 (20) | 3 (15) | 0 (0) | 0·016 | 0·032 |
| No | 12 (80) | 17 (85) | 41 (100) | ||
Data are n (%). aGroup A, n =14; group B, n =18 and group C, n =37; bgroup C, n =39; cgroup C, n =40; dgroup B, n =19 and group C, n =40.
Correlation analysis of carriers of two IL36RN mutations or one to two IL36RN mutations vs. noncarriers in 61 patients with generalized pustular psoriasis (GPP)
| Category and subcategory | Group A, carriers of two IL36RN mutations (n =15) | Group B, carriers of one to two IL36RN mutations (n =20) | Group C, noncarriers of IL36RN mutation (n =41) | P‐value (A vs. C) | P‐value (B vs. C) |
| Sex | |||||
| Male | 4 (27) | 5 (25) | 13 (32) | 1 | 0·76 |
| Female | 11 (73) | 15 (75) | 28 (68) | ||
| Course of diseasea | |||||
| Episodic | 13 (93) | 15 (83) | 26 (70) | 0·14 | 0·34 |
| Continuous | 1 (7) | 3 (17) | 11 (30) | ||
| Palmoplantar pustular psoriasis | |||||
| Yes | 3 (20) | 5 (25) | 4 (10) | 0·36 | 0·13 |
| No | 12 (80) | 15 (75) | 37 (90) | ||
| Tongue affectedb | |||||
| Yes | 4 (27) | 5 (25) | 8 (21) | 0·71 | 0·74 |
| No | 11 (73) | 15 (75) | 31 (79) | ||
| Psoriasis vulgarisc | |||||
| Yes | 3 (20) | 5 (25) | 20 (50) | 0·066 | 0·095 |
| No | 12 (80) | 15 (75) | 20 (50) | ||
| Nails affected | |||||
| Yes | 5 (33) | 5 (25) | 11 (27) | 0·74 | 1 |
| No | 10 (67) | 15 (75) | 30 (73) | ||
| Joints affected | |||||
| Yes | 4 (27) | 6 (30) | 12 (29) | 1 | 1 |
| No | 11 (73) | 14 (70) | 29 (71) | ||
| Cholangitis | |||||
| Yes | 1 (7) | 1 (5) | 1 (2) | 0·46 | 1 |
| No | 14 (93) | 19 (95) | 40 (98) | ||
| Family history positived | |||||
| Yes | 9 (60) | 11 (58) | 17 (43) | 0·36 | 0·4 |
| No | 6 (40) | 8 (42) | 23 (58) | ||
| Consanguinity | |||||
| Yes | 3 (20) | 3 (15) | 0 (0) | 0·016 | 0·032 |
| No | 12 (80) | 17 (85) | 41 (100) | ||
| Category and subcategory | Group A, carriers of two IL36RN mutations (n =15) | Group B, carriers of one to two IL36RN mutations (n =20) | Group C, noncarriers of IL36RN mutation (n =41) | P‐value (A vs. C) | P‐value (B vs. C) |
| Sex | |||||
| Male | 4 (27) | 5 (25) | 13 (32) | 1 | 0·76 |
| Female | 11 (73) | 15 (75) | 28 (68) | ||
| Course of diseasea | |||||
| Episodic | 13 (93) | 15 (83) | 26 (70) | 0·14 | 0·34 |
| Continuous | 1 (7) | 3 (17) | 11 (30) | ||
| Palmoplantar pustular psoriasis | |||||
| Yes | 3 (20) | 5 (25) | 4 (10) | 0·36 | 0·13 |
| No | 12 (80) | 15 (75) | 37 (90) | ||
| Tongue affectedb | |||||
| Yes | 4 (27) | 5 (25) | 8 (21) | 0·71 | 0·74 |
| No | 11 (73) | 15 (75) | 31 (79) | ||
| Psoriasis vulgarisc | |||||
| Yes | 3 (20) | 5 (25) | 20 (50) | 0·066 | 0·095 |
| No | 12 (80) | 15 (75) | 20 (50) | ||
| Nails affected | |||||
| Yes | 5 (33) | 5 (25) | 11 (27) | 0·74 | 1 |
| No | 10 (67) | 15 (75) | 30 (73) | ||
| Joints affected | |||||
| Yes | 4 (27) | 6 (30) | 12 (29) | 1 | 1 |
| No | 11 (73) | 14 (70) | 29 (71) | ||
| Cholangitis | |||||
| Yes | 1 (7) | 1 (5) | 1 (2) | 0·46 | 1 |
| No | 14 (93) | 19 (95) | 40 (98) | ||
| Family history positived | |||||
| Yes | 9 (60) | 11 (58) | 17 (43) | 0·36 | 0·4 |
| No | 6 (40) | 8 (42) | 23 (58) | ||
| Consanguinity | |||||
| Yes | 3 (20) | 3 (15) | 0 (0) | 0·016 | 0·032 |
| No | 12 (80) | 17 (85) | 41 (100) | ||
Data are n (%). aGroup A, n =14; group B, n =18 and group C, n =37; bgroup C, n =39; cgroup C, n =40; dgroup B, n =19 and group C, n =40.
Discussion
In the majority of patients with GPP, we did not identify a causal variant, indicating a role of further disease‐causing/disease‐contributing genes. To the best of our knowledge, a comprehensive analysis of the three genes has not been reported in larger patient groups of patients with pustular psoriasis. With regard to the genes AP1S3 and CARD14, our results suggest their variants have a much lower impact in GPP than variants in IL36RN.
Of note, in the currently largest European control cohort, the sum of the allele frequencies of the two AP1S3 missense variants c.11T>G (p.Phe4Cys) and c.97C>T (p.Arg33Trp) was 1% higher (2·3% in 32 864 Non‐Finnish European individuals in the Exome Aggregation Consortium) than described in the initial study by Setta‐Kaffetzi et al.6 (1·3% in 1695 control exomes), possibly explaining why associations of these variants had been previously identified. All current studies performed for the AP1S3 gene are underpowered to detect associations in this frequency range, but more importantly, the study by Setta‐Kaffetzi et al.6 followed by the one of Mahil et al.16 indicated that those variants were functional. Nevertheless, those variants have been described as less penetrant susceptibility alleles in contrast to the mutations in IL36RN. Our finding of two heterozygous carriers of AP1S3 disease‐contributing variants within the small group of only two patients with AGEP is an interesting one and might hint to be a relevant variant in a broader disease spectrum, although it needs further confirmation within a larger patient cohort to be a significant one.
Apart from a missense variant in mainly Asian cohorts of patients with GPP,12,15 variants in CARD14 were relevant in single patients with GPP and were overall associated, but do not seem to play a major role in the group of European patients. In addition, the more recent interpretation of a single missense variant in CARD1412 renders our previous association of PPP with missense variants in CARD148 a nonsignificant one.
As recently reported,3 carriers of IL36RN mutations were significantly younger at age of onset than noncarriers. In disagreement with previous studies,3,15 frequencies of patients with concomitant psoriasis vulgaris were comparable between the two groups. Although considerable additional clinical data were collected, no associations of IL36RN with other clinical characteristics were observed.
When sequencing noncoding regions of IL36RN, no evidence for additional noncoding mutations in carriers of a single heterozygous IL36RN mutation was found. Also, the quantitative analysis rendered an intragenic deletion or duplication on the second allele unlikely. The frequency of heterozygous carriers of IL36RN mutations (8%) is much higher (15 times) than the frequency of heterozygous carriers in the general population (0·54%). Additionally, none of the parents of our GPP patients, carrying a heterozygous IL36RN mutation, had pustular skin disease. These findings suggest a contributing effect of the IL36RN mutations in heterozygous patients with pustular psoriasis, and a more complex, probably oligogenic inheritance. An oligogenic rather than a purely monogenic inheritance is further supported by IL36RN mutations occurring with additional variants in other genes in 15% of those carriers. Previously, epistatic effects of IL36RN and AP1S3 have been suggested in a single European patient carrying one IL36RN mutation and one of the two missense variants in AP1S3.16 To our knowledge, more comprehensive studies in larger study groups have not been performed. The oligogenic basis of GPP might currently be underestimated, as our study suggests that genetic risk factors other than IL36RN mutations remain to be identified in the majority of patients.
In conclusion, our systematic analysis of genotype–phenotype correlations suggests a very significant correlation of IL36RN mutations with a younger age at disease onset, with no further significant correlations observed. Patients with GPP, with IL36RN mutations, carried an additional rare variant in either CARD14 or AP1S3 in 15% of cases, indicating a more complex inheritance. Additional genetic factors outside IL36RN probably contribute to the pathogenesis of pustular skin disease in heterozygous IL36RN mutation carriers.
Acknowledgments
We are grateful to all patients and their parents for participation in this study. We thank Anne Gerschütz, Tim Aberle and Jessica Welss for excellent technical assistance. The study was partly supported by grants to U.H. from the German Research Foundation (DFG 2163/1‐1; CRC1181 – project A05), from the Interdisciplinary Centre for Clinical Research (laboratory rotation) of the Clinical Center Erlangen of the Friedrich‐Alexander‐Universität Erlangen‐Nürnberg, Germany, and from the Bundesministerium für Bildung und Forschung (BMBF Metarthros 01EC1407A).
References
Author notes
Funding sources The study was partly supported by grants to U.H. from the German Research Foundation (DFG 2163/1‐1; CRC1181 – project A05), from the Interdisciplinary Centre for Clinical Research (laboratory rotation) of the Clinical Center Erlangen of the Friedrich‐Alexander‐Universität Erlangen‐Nürnberg, Germany, and from the Bundesministerium für Bildung und Forschung (BMBF Metarthros 01EC1407A).
Conflicts of interest R.M. has been an advisor and/or received speakers’ honoraria and/or received grants and/or participated in clinical trials or received travel support from the following companies: AbbVie, Biogen IDEC GmbH, Celgene, Janssen‐Cilag GmbH, LEO Pharma GmbH, Lilly, Merck Serono GmbH, Merck Sharp & Dohme GmbH, Novartis Pharma GmbH und Pfizer GmbH. D.W.‐T. has been an advisor and/or received speakers’ honoraria or travel expense reimbursements and/or received grants from and/or participated in clinical trials of: AbbVie, Almirall, Amgen, Biogen, Boehringer Ingelheim Pharma, Celgene, Forward Pharma, GlaxoSmithKline, Janssen‐Cilag, LEO Pharma, Lilly, Medac, Merck Sharp & Dohme, Novartis, Pfizer, UCB and VBL. V.O. received reimbursements for talks and/or travels from the companies Janssen‐Cilag, AbbVie, Biogen, Celgene, UCB, Novartis and Pfizer, and is principal investigator in the study CC‐10004‐PPSO‐001 (Celgene). K.S. has been an advisor and/or received speakers’ honoraria or travel expense reimbursements and/or received grants from and/or participated in clinical trials of the companies AbbVie, Amgen, Biogen, Boehringer Ingelheim, Celgene, Janssen‐Cilag, Lilly, Merck Sharp & Dohme, Novartis and Pfizer. K.R. was a member of the advisory board and/or was a speaker and/or involved in faculty education and/or was an author and/or was involved in research and/or in clinical studies and/or has a patent and/or is stockholder and/or consults for the following companies: AbbVie, Amgen, Biogen, Boehringer Ingelheim, Celgene, Covagen, Forward Pharma, GlaxoSmithKline, Janssen‐Cilag, LEO Pharma, Lilly, Medac, Merck Sharp & Dohme, Novartis, Pfizer, Regeneron, Takeda, UCB, Xenoport and SCIderm. M.S. was scientifically supported by Pfizer and Novartis and is a member of the advisory boards of AbbVie, Celgene, Janssen‐Cilag, Lilly, Pfizer, MSD, Novartis, Amgen, LEO Pharma and Actelion; he received speakers’ honoraria from AbbVie, Actelion, Janssen‐Cilag, LEO Pharma, MSD, Novartis and Pfizer and was involved in the clinical studies of AbbVie, Actelion, Amgen, Galderma, Janssen‐Cilag, Pfizer and Regeneron.
R.M., D.W.‐T., V.O., P.G. and S.L. contributed equally to this work.

{kind=link}
{kind=link}