





Abstract
Mitochondrial calcium-sensitive potassium (mKCa) channels are involved in cardiac preconditioning. In the present study, we investigated whether also ischaemic-, morphine-induced post-conditioning, or both is mediated by the activation of mKCa channels in the rat heart in vitro.
Animals were treated in compliance with institutional and national guidelines. Male Wistar rats were randomly assigned to one of seven groups (each n=7). Control animals were not further treated. Post-conditioning was induced either by 3×30 s of ischaemia/reperfusion (I-PostC) or by administration of morphine (M-PostC, 1 µM) for 15 min at the onset of reperfusion. The mKCa-channel inhibitor paxilline (1 µM) was given with and without post-conditioning interventions (M-PostC+Pax, I-PostC+Pax, and Pax). As a positive control, we determined whether direct activation of mKCa channels with NS1619 (10 µM) induced cardiac post-conditioning (NS1619). Isolated hearts underwent 35 min ischaemia followed by 120 min reperfusion. At the end of reperfusion, infarct sizes were measured by triphenyltetrazolium chloride staining.
In the control group, infarct size was 53 (5)% of the area at risk. Morphine- and ischaemic post-conditioning reduced infarct size in the same range [M-PostC: 37 (4)%, I-PostC: 35 (5)%; each P<0.05 vs control]. The mKCa-channel inhibitor paxilline completely blocked post-conditioning [M-PostC+Pax: 47 (7)%, I-PostC+Pax: 51 (3)%; each P<0.05 vs M-PostC and I-PostC, respectively]. Paxilline itself had no effect on infarct size (NS vs control). NS1619 reduced infarct size to 33 (4)% (P<0.05 vs control).
Ischaemic- and morphine-induced post-conditioning is mediated by the activation of mKCa channels.
The heart can be protected from an ischaemic insult by short exposure to ischaemia or morphine before or after the ischaemic insult.
By using an inhibitor, these authors showed that like preconditioning, post-conditioning occurs via the activation of calcium-sensitive potassium channels in mitochondria.
An important role for mitochondria in cardioprotection is shown.
Pre- and post-conditioning are potent measures to protect the heart against the consequences of ischaemia and reperfusion.1 Cardioprotection by pre- and post-conditioning can be induced by various stimuli, for example, brief cycles of ischaemia,2,3 but also pharmacologically, for example, with morphine.4,5 The mechanisms by which opioids protect the myocardium are common to ischaemic preconditioning. The opening of mitochondrial ATP-sensitive potassium (mKATP) channels, which are involved in the regulation of mitochondrial functions, is a key step mediating the cardioprotective effects of both ischaemic- and morphine-induced preconditioning. Next to the activation of mKATP channels,6 preconditioning seems to be mediated by another class of K+ channels, the mitochondrial calcium-sensitive potassium (mKCa) channel.7,8 In a recent study, Cao and colleagues9 showed that mKCa channels are involved in ischaemic preconditioning. Furthermore, these authors demonstrated that the activation of KCa channels confer cardioprotection independently of mKATP channels and vice versa.9 Nothing is known about the involvement of KCa channels in ischaemic- and morphine-induced post-conditioning.
Here, we hypothesize that ischaemic- and morphine-induced post-conditioning is, as preconditioning, mediated by the activation of KCa channels.
Methods
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and approved by the Institutional Committee for Animal Care and Use (Academic Medical Centre Amsterdam, The Netherlands).
Chemicals and reagents
Morphine–HCl was purchased from Centrafarm (Etten-Leur, The Netherlands). All other chemicals were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands).
Surgical preparation
Male Wistar rats (Charles River, The Netherlands) were used for these studies. Rats were maintained on a 12:12 light/dark schedule (lights on at 06:00 h) with food and water provided ad libitum. The rats were acclimatized to the local animal facility for at least 7 days before the experiment. Rats were anaesthetized with S-ketamine (150 mg kg−1 i.p.). Before starting surgery, adequate depth of anaesthesia was verified by the absence of reaction after painful stimuli. After thoracotomy, hearts were quickly excised and mounted on a Langendorff system and were perfused at constant pressure (80 mm Hg) with the Krebs–Henseleit solution containing (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 0.5 EDTA, 2.25 CaCl2, 11 glucose, 1 lactate, and 0.1 pyruvate at 37 °C. A fluid-filled balloon was inserted into the left ventricle and end-diastolic pressure was set at 1–4 mm Hg. All hearts underwent a stabilization period of 20 min. Heart rate, myocardial function (isovolumetric left ventricular pressure), coronary flow, left ventricular end-diastolic pressure, and dP/dtmax were measured continuously. The time of maximal ischaemic contracture and the level of maximal ischaemic contracture were determined in each experiment by checking the course of contracture development during index ischaemia and by selecting the time point (measured from the onset of index ischaemia) when the contracture reached its highest level. Arrhythmic intervals were not used for data analysis. The rate–pressure product was calculated as heart rate×(maximal left ventricular pressure−left ventricular end-diastolic pressure).
Experimental design
Hearts were assigned to one of the seven experimental groups (each n=7, Fig. 1). Hearts from all groups underwent 35 min of global ischaemia by stopping coronary perfusion (no-flow ischaemia), followed by 120 min of reperfusion. In the control group, hearts underwent no further intervention. Ischaemic post-conditioning (I-PostC) was induced by three cycles of 30 s ischaemia/reperfusion at the onset of reperfusion.3,10 To investigate whether morphine induces post-conditioning, 1 µM morphine–HCl11 (M-PostC) was given over 15 min starting with the reperfusion period. To test whether the cardioprotective effects are mediated by mKCa-channel activation, we combined both the groups with the mKCa-channel inhibitor paxilline. The concentration of 1 µM9,12 was chosen based on the current literature (I-PostC+Pax, M-PostC+Pax). Paxilline was also given without any other treatment (Pax). As a positive control group, we determined whether direct activation of mKCa channels with NS1619 (10 µM)9 induces cardiac post-conditioning (NS1619). Morphine was dissolved in NaCl (0.9%) and paxilline and NS1619 were dissolved in DMSO and separately infused into a mixing chamber placed in the perfusion system.
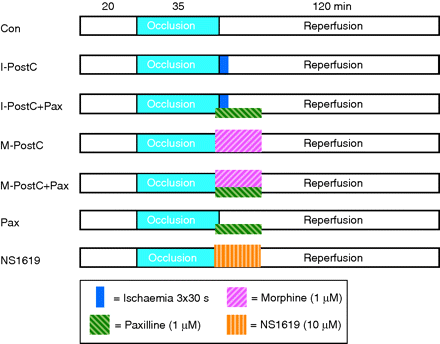
Experimental protocol. Con, control; I-PostC, ischaemic post-conditioning; M-PostC, morphine post-conditioning; Pax, paxilline; NS1619, mKCa-channel activator.
At 120 min of reperfusion, hearts were frozen at −20°C. Subsequently, infarct sizes were determined by triphenyltetrazolium chloride staining. Therefore, heart slices (7–9 per heart) were incubated with 0.75% triphenyltetrazolium chloride solution for 10 min at 37°C and fixed in 4% formalin solution for 24 h at room temperature. The infarcted area was determined by planimetry using SigmaScan Pro 5® computer software (SPSS Science Software, Chicago, IL, USA) by two independent researchers in a blinded manner.
Statistical analysis
The sample size was calculated using GraphPad StatMate™ Version 1.01 (GraphPad Software, San Diego, CA, USA). Sample size analysis revealed a group size of n=7 was necessary to detect a difference in infarct size of 15% with a power of 80% and an α<0.05. Data are expressed as mean and standard deviation (sd), or median with 25th and 75th percentiles. Statistical analysis (SPSS Science Software, version 17.0) of the haemodynamic variables was performed by the Kruskal–Wallis H-test for time and treatment (experimental group) effects. If an overall significance was found, comparisons between groups were made by the Student–Newman–Keuls post hoc test. Infarct sizes were analysed by Kruskal–Wallis H-test followed by the Student–Newman–Keuls post hoc test. Changes within and between groups were considered statistically significant if P<0.05.
Results
No differences in body or heart weight were observed between the groups (Table 1).
Weights and ischaemic contracture. Data are mean (sd). Con, control; I-PostC, ischaemic post-conditioning; M-PostC, morphine post-conditioning; Pax, paxilline; NS1619, mKCa channel activator
| Body weight (g) | Heart weight (g) | Time of max. ischaemic contracture (min) | Level of max. ischaemic contracture (mm Hg) | ||
|---|---|---|---|---|---|
| Wet | Dry | ||||
| Con | 366 (20) | 1.5 (0.1) | 0.2 (0.02) | 21 (1) | 78 (7) |
| I-PostC | 367 (17) | 1.4 (0.2) | 0.2 (0.04) | 19 (2) | 82 (7) |
| I-PostC+Pax | 371 (30) | 1.3 (0.2) | 0.2 (0.03) | 18 (2) | 82 (6) |
| M-PostC | 373 (26) | 1.4 (0.1) | 0.2 (0.02) | 19 (1) | 83 (5) |
| M-PostC+Pax | 365 (30) | 1.4 (0.1) | 0.2 (0.04) | 20 (2) | 83 (6) |
| Pax | 377 (33) | 1.3 (0.1) | 0.2 (0.02) | 19 (1) | 78 (6) |
| NS1619 | 366 (26) | 1.4 (0.1) | 0.2 (0.02) | 20 (2) | 76 (3) |
| Body weight (g) | Heart weight (g) | Time of max. ischaemic contracture (min) | Level of max. ischaemic contracture (mm Hg) | ||
|---|---|---|---|---|---|
| Wet | Dry | ||||
| Con | 366 (20) | 1.5 (0.1) | 0.2 (0.02) | 21 (1) | 78 (7) |
| I-PostC | 367 (17) | 1.4 (0.2) | 0.2 (0.04) | 19 (2) | 82 (7) |
| I-PostC+Pax | 371 (30) | 1.3 (0.2) | 0.2 (0.03) | 18 (2) | 82 (6) |
| M-PostC | 373 (26) | 1.4 (0.1) | 0.2 (0.02) | 19 (1) | 83 (5) |
| M-PostC+Pax | 365 (30) | 1.4 (0.1) | 0.2 (0.04) | 20 (2) | 83 (6) |
| Pax | 377 (33) | 1.3 (0.1) | 0.2 (0.02) | 19 (1) | 78 (6) |
| NS1619 | 366 (26) | 1.4 (0.1) | 0.2 (0.02) | 20 (2) | 76 (3) |
Weights and ischaemic contracture. Data are mean (sd). Con, control; I-PostC, ischaemic post-conditioning; M-PostC, morphine post-conditioning; Pax, paxilline; NS1619, mKCa channel activator
| Body weight (g) | Heart weight (g) | Time of max. ischaemic contracture (min) | Level of max. ischaemic contracture (mm Hg) | ||
|---|---|---|---|---|---|
| Wet | Dry | ||||
| Con | 366 (20) | 1.5 (0.1) | 0.2 (0.02) | 21 (1) | 78 (7) |
| I-PostC | 367 (17) | 1.4 (0.2) | 0.2 (0.04) | 19 (2) | 82 (7) |
| I-PostC+Pax | 371 (30) | 1.3 (0.2) | 0.2 (0.03) | 18 (2) | 82 (6) |
| M-PostC | 373 (26) | 1.4 (0.1) | 0.2 (0.02) | 19 (1) | 83 (5) |
| M-PostC+Pax | 365 (30) | 1.4 (0.1) | 0.2 (0.04) | 20 (2) | 83 (6) |
| Pax | 377 (33) | 1.3 (0.1) | 0.2 (0.02) | 19 (1) | 78 (6) |
| NS1619 | 366 (26) | 1.4 (0.1) | 0.2 (0.02) | 20 (2) | 76 (3) |
| Body weight (g) | Heart weight (g) | Time of max. ischaemic contracture (min) | Level of max. ischaemic contracture (mm Hg) | ||
|---|---|---|---|---|---|
| Wet | Dry | ||||
| Con | 366 (20) | 1.5 (0.1) | 0.2 (0.02) | 21 (1) | 78 (7) |
| I-PostC | 367 (17) | 1.4 (0.2) | 0.2 (0.04) | 19 (2) | 82 (7) |
| I-PostC+Pax | 371 (30) | 1.3 (0.2) | 0.2 (0.03) | 18 (2) | 82 (6) |
| M-PostC | 373 (26) | 1.4 (0.1) | 0.2 (0.02) | 19 (1) | 83 (5) |
| M-PostC+Pax | 365 (30) | 1.4 (0.1) | 0.2 (0.04) | 20 (2) | 83 (6) |
| Pax | 377 (33) | 1.3 (0.1) | 0.2 (0.02) | 19 (1) | 78 (6) |
| NS1619 | 366 (26) | 1.4 (0.1) | 0.2 (0.02) | 20 (2) | 76 (3) |
Infarct size measurement
In the control group, infarct size was 53 (5)% (Fig. 2). Ischaemic- and morphine-induced post-conditioning reduced infarct size in the same range [I-PostC: 35 (5)%, M-PostC: 37 (4)%; each P<0.05 vs control; Fig. 2]. Both effects were blocked by the mKCa-channel inhibitor paxilline [I-PostC+Pax: 51 (3)%, M-PostC+Pax: 47 (7)%; each P<0.05 vs I-PostC and M-PostC, respectively; Fig. 2], while paxilline itself had no effect on infarct size [Pax: 48 (4)%, NS vs control]. The activation of mKCa channels with NS1619 also reduced infarct size to 33 (4)% [P<0.05 vs control; Fig. 2].
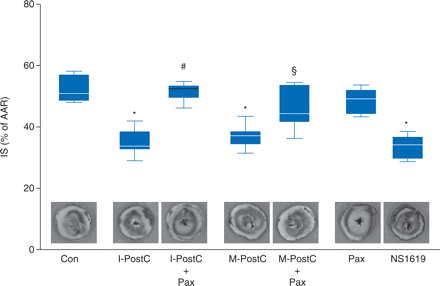
Infarct size data. Histogram shows the infarct size (% of area at risk, AAR) of controls (Con), ischaemic post-conditioning (I-PostC), ischaemic post-conditioning and paxilline (I-PostC+Pax), morphine post-conditioning (M-PostC), morphine post-conditioning and paxilline (M-PostC+Pax), paxilline alone (Pax), and NS1619. The boxes represent the lower (25th) and upper (75th) quartiles, the horizontal line represents the mean, the whiskers extend to 95% confidence intervals for the mean, *P<0.05 vs Con, #P<0.05 vs I-PostC, and §P<0.05 vs M-PostC.
Haemodynamic variables
No significant differences in haemodynamics were observed between the experimental groups during baseline, ischaemia, and reperfusion (Supplementary Table S1).
Discussion
The main finding of our study is that both ischaemic- and morphine-induced post-conditioning is mediated by the activation of mKCa channels.
Stimulation of opioid receptors enhances the resistance of the myocardium against ischaemia and reperfusion injury.13–16 Schultz and colleagues showed that the non-selective opioid receptor antagonist naloxone abrogated the cardioprotective effect of ischaemic preconditioning17 and, furthermore, that morphine initiates cardioprotection.4 In a later study, these authors also demonstrated that the cardioprotective effect of ischaemic preconditioning was mediated by δ1-opioid receptor activation.18 Evidence suggests that also morphine-induced preconditioning is mediated via the activation of δ-opioid receptors.19 Furthermore, it was shown that the regulation of mitochondrial function by the activation of mitochondrial ATP-sensitive potassium (mKATP) channels plays a central role in both morphine-induced20–22 and ischaemic-induced cardioprotection.23 Mitochondria are involved in the putative end-effector mechanism of pre- and post-conditioning. A possible candidate as end-effector of cardioprotection against ischaemia and reperfusion injury is the mitochondrial permeability transition pore (mPTP).24–26
Next to mKATP channels, cardioprotection seems to be mediated by another class of K+ channels, the calcium-sensitive potassium (KCa) channel.7,8 A central role of KCa channels in preconditioning has been shown in several studies as the pharmacological activation of these channels initiates cardioprotection, and pharmacological preconditioning can be blocked by KCa channel antagonists.7–9,27,28 In 2002, Xu and colleagues8 substantiated the existence of KCa channels in the inner mitochondrial membrane of ventricular myocytes and reported a cardioprotective effect via mKCa channel activation. Cao and colleagues9 showed that ischaemic preconditioning is triggered by the activation of mKCa channels and abolished by the mKCa channel inhibitor paxilline. Direct activation of mKCa channels with the mKCa channel activator NS16198 reduced the infarct size in the same range as ischaemic preconditioning.9 In a recent study, we demonstrated that preconditioning did not only reduce infarct size by the activation of mKCa channels, but it also caused a significant reduction in the mitochondrial respiratory control index.28 Furthermore, the reduction in the respiratory control index was completely abolished by co-administration of the mKCa channel blocker iberiotoxin and we concluded that cardioprotection is mediated by the activation of mKCa channels leading to mild mitochondrial uncoupling. Mild mitochondrial uncoupling during the trigger phase of preconditioning may represent a common characteristic of mitochondria in a ‘conditioned’ state.8,29–31 There is no study available investigating the involvement of mKCa channels in ischaemic-, morphine-induced post-conditioning, or both.
In the present study, ischaemic- and morphine-induced post-conditioning reduced the infarct size in the same range. Both effects were completely abrogated by paxilline, supporting our hypothesis that mKCa channels are involved in both forms of cardioprotection. Furthermore, post-conditioning by direct activation of mKCa channels with NS1619 induced infarct size reduction. In the present study, paxilline was used to block mKCa channels for several reasons: (i) paxilline was often used in Langendorff perfusion studies investigating preconditioning signalling mechanisms,9,12,32,33 (ii) there is evidence that paxilline does not act on mKATP channels because paxilline did not prevent preconditioning induced by the mKATP channel activator diazoxide,9 and (iii) we previously showed that paxilline is capable of abolishing the opening of mKCa channels in isolated mitochondria.34 Besides paxilline, there are not many other mKCa channel blockers available, for example, charybdotoxin or iberiotoxin. Iberiotoxin, for example, is an mKCa channel blocker also suitable for in vivo use,28 whereas paxilline is predominantly used for in vitro experiments. However, we did not check whether iberiotoxin or charybdotoxin is also able to abolish ischaemic- and morphine-induced post-conditioning. The opening of mKCa channels is capable of causing a slight increase in mitochondrial reactive oxygen species generation.35 Stowe and colleagues27 demonstrated that the cardioprotective effect of the mKCa channel agonist NS1619 requires superoxide radical generation during the preconditioning stimulus. Furthermore, these authors demonstrated that cardioprotection by NS1619 reduces mitochondrial calcium overload and mitochondrial reactive oxygen species production during the subsequent period of ischaemia and early reperfusion.27 Such a reduction in mitochondrial calcium overload and reactive oxygen species generation has been suggested to prevent mPTP opening.36,37 There is evidence that the mKCa channel is located upstream of the mPTP, because cardioprotection induced by the activation of mKCa channels was abolished by the opening of the mPTP. Vice versa, the inhibition of the mKCa channel with paxilline did not block protection induced by the inhibition of the mPTP with cyclosporine A.9 Whether cardiac post-conditioning is also mediated by the regulation of the mPTP due to mKCa channel activation is unknown.
The results of the present study have to be interpreted within the scope of some limitations. First, possible upstream mechanisms of mKCa channel activation remain elusive. In 2005, Weihrauch and colleagues38 demonstrated that morphine-induced post-conditioning involves phosphatidylinositol-3-kinase (PI3K). An involvement of PI3K was also shown for ischaemic post-conditioning.39 Beside PI3K, glycogen synthase kinase-3β and Akt were also shown to be involved in morphine post-conditioning.40 Opioid receptors are G-protein-coupled receptors whose activation inhibits adenylate cyclase. The post-receptor signalling after opioid receptor activation has not completely been defined. We cannot rule out that morphine confers preconditioning also via intracellular pathways leading to the activation of mKCa channels. It has been shown that the regulation of mitochondrial function by the activation of mKATP channels plays a central role in morphine-induced cardioprotection.20,21 Ludwig and colleagues demonstrated that morphine-enhanced isoflurane induced preconditioning via the activation of mKATP channels.41 The involvement of mitochondria in morphine-induced cardioprotection is supported by data from our group, showing that morphine causes a translocation of hexokinase to the mitochondria.42 However, it is unknown how morphine regulates mKCa channel activation to induce preconditioning. KCa channels can be regulated via enzymatic pathways, that is, by protein kinase A activation.43,44 It has been shown that mKCa channel activation is involved in desflurane preconditioning and that protein kinase A is located upstream of the mKCa channel.45 The activity of protein kinase A as a possible upstream activator of KCa channels depends on the cellular level of cAMP (PKA is known as cAMP-dependent protein kinase). It was shown that tissue cAMP levels were increased after morphine treatment,46 but it is unknown whether morphine increases cAMP levels by increasing cAMP production or by decreasing cAMP decomposition, that is, stimulation of adenylyl cyclase or inhibition of phosphodiesterase. However, in the present study, we did not investigate whether these enzymes are related to mKCa channel activation. Secondly, Obame and colleagues47 showed recently that morphine post-conditioning is associated with an increased mitochondrial resistance and the inhibition of mPTP opening that is related to PI3K. In the present study, we demonstrate that both ischaemic and morphine post-conditioning are abolished by pharmacological mKCa-channel blockade. However, we did not investigate a direct effect of ischaemic and morphine post-conditioning on mitochondria, for example, by analysing mitochondrial respiration, redox potential, or ‘stress’-induced mPTP opening. Thirdly, the present study was designed to elucidate the involvement of mKCa channels in both ischaemic- and morphine-induced post-conditioning. Therefore, a group receiving the mKCa-channel activator NS1619 was included as a positive control to demonstrate that mKCa-channel activation after ischaemia is capable of inducing post-conditioning. However, there is an ongoing debate whether NS1619 specifically activates mKCa channels or whether NS1619 has other non-mitochondrial side-effects or effects on mitochondrial function that are not due to mKCa-channel activation.48–51 From the data of the present study, we cannot completely exclude the possibility that the infarct size-reducing effect of NS1619 is due to other mechanisms than the activation of mKCa channels.
In summary, this is the first study demonstrating that brief cycles of ischaemia and morphine initiate post-conditioning via the activation of mKCa channels in the isolated rat heart.
Supplementary material
Supplementary material is available at British Journal of Anaesthesia online.
Conflict of interest
None declared.
Funding
This study was supported by institutional and departmental sources.



{kind=link}
{kind=link}