





Abstract
Severe sepsis and septic shock is common and frequently fatal. Over the last few years, the primary treatments demonstrated to improve outcome from several major clinical trials have finally emerged. However, translating these recent therapeutic advances to routine clinical practice has proven controversial, and new approaches of additional strategies are continued to be developed. Given their pleiotropic effects related to many pathophysiological determinants of sepsis, statin therapy could be the next step in the search for adjuvant therapy. A future challenge may be to test both the efficacy and the safety by large randomized controlled clinical trials ascertaining the effects of statins administered at the onset of sepsis and in patients with severe sepsis or septic shock admitted into intensive care units.
The importance of sepsis
The pathophysiological process of sepsis is a disease continuum from infection, signs and symptoms of infection (SSI, used to be termed as systemic inflammatory response syndrome SIRS), sepsis, severe sepsis, and multi-organ dysfunction.54,71 Internationally recognized definitions are outlined in Table 1.
Sepsis: terminology and definitions
| Infection | A pathologic process caused by the invasion of normally sterile tissue or fluid or body cavity by pathogenic or potentially pathogenic microorganisms |
| Signs and symptoms of infection | Requires any two of the following signs and symptoms both present and new to the patient: temperature >38.3°C or <36.0°C; heart rate >90 beat min−1; respiratory rate >20 bpm; acutely altered mental status; leucocytosis—WBC >12 000 μl−1; leucopaenia—WBC <4000 μl−1; hyperglycaemia (plasma glucose >7.7 mmol litre-1) in the absence of diabetes; plasma C-reactive protein >2 sd above the normal value |
| Sepsis | The clinical syndrome defined by the presence of both signs and symptoms of infection as described above and documented or suspected new infection |
| Severe sepsis | Sepsis complicated by one organ dysfunction |
| Multi-organ failure | Two or more organ dysfunction |
| Organ dysfunction | Defined using the definitions used for the Sequential Organ Failure Assessment (SOFA) score:40Pao2/Fio2≤26.7 kPa; noradrenaline/adrenaline ≥0.1 μg kg−1 min−1; creatinine ≥330 μmol litre−1; platelets ≤50×103 ml−1; bilirubin ≥102 μmol litre−1; Glasgow Coma Score <9 |
| Septic shock | In adults applies to a state of acute circulatory failure unexplained by other causes, defined as persistent arterial hypotension [a systolic arterial pressure (SBP) <90, MAP<60, or a reduction in SBP 40 mm Hg from baseline despite adequate volume resuscitation] |
| Infection | A pathologic process caused by the invasion of normally sterile tissue or fluid or body cavity by pathogenic or potentially pathogenic microorganisms |
| Signs and symptoms of infection | Requires any two of the following signs and symptoms both present and new to the patient: temperature >38.3°C or <36.0°C; heart rate >90 beat min−1; respiratory rate >20 bpm; acutely altered mental status; leucocytosis—WBC >12 000 μl−1; leucopaenia—WBC <4000 μl−1; hyperglycaemia (plasma glucose >7.7 mmol litre-1) in the absence of diabetes; plasma C-reactive protein >2 sd above the normal value |
| Sepsis | The clinical syndrome defined by the presence of both signs and symptoms of infection as described above and documented or suspected new infection |
| Severe sepsis | Sepsis complicated by one organ dysfunction |
| Multi-organ failure | Two or more organ dysfunction |
| Organ dysfunction | Defined using the definitions used for the Sequential Organ Failure Assessment (SOFA) score:40Pao2/Fio2≤26.7 kPa; noradrenaline/adrenaline ≥0.1 μg kg−1 min−1; creatinine ≥330 μmol litre−1; platelets ≤50×103 ml−1; bilirubin ≥102 μmol litre−1; Glasgow Coma Score <9 |
| Septic shock | In adults applies to a state of acute circulatory failure unexplained by other causes, defined as persistent arterial hypotension [a systolic arterial pressure (SBP) <90, MAP<60, or a reduction in SBP 40 mm Hg from baseline despite adequate volume resuscitation] |
Sepsis: terminology and definitions
| Infection | A pathologic process caused by the invasion of normally sterile tissue or fluid or body cavity by pathogenic or potentially pathogenic microorganisms |
| Signs and symptoms of infection | Requires any two of the following signs and symptoms both present and new to the patient: temperature >38.3°C or <36.0°C; heart rate >90 beat min−1; respiratory rate >20 bpm; acutely altered mental status; leucocytosis—WBC >12 000 μl−1; leucopaenia—WBC <4000 μl−1; hyperglycaemia (plasma glucose >7.7 mmol litre-1) in the absence of diabetes; plasma C-reactive protein >2 sd above the normal value |
| Sepsis | The clinical syndrome defined by the presence of both signs and symptoms of infection as described above and documented or suspected new infection |
| Severe sepsis | Sepsis complicated by one organ dysfunction |
| Multi-organ failure | Two or more organ dysfunction |
| Organ dysfunction | Defined using the definitions used for the Sequential Organ Failure Assessment (SOFA) score:40Pao2/Fio2≤26.7 kPa; noradrenaline/adrenaline ≥0.1 μg kg−1 min−1; creatinine ≥330 μmol litre−1; platelets ≤50×103 ml−1; bilirubin ≥102 μmol litre−1; Glasgow Coma Score <9 |
| Septic shock | In adults applies to a state of acute circulatory failure unexplained by other causes, defined as persistent arterial hypotension [a systolic arterial pressure (SBP) <90, MAP<60, or a reduction in SBP 40 mm Hg from baseline despite adequate volume resuscitation] |
| Infection | A pathologic process caused by the invasion of normally sterile tissue or fluid or body cavity by pathogenic or potentially pathogenic microorganisms |
| Signs and symptoms of infection | Requires any two of the following signs and symptoms both present and new to the patient: temperature >38.3°C or <36.0°C; heart rate >90 beat min−1; respiratory rate >20 bpm; acutely altered mental status; leucocytosis—WBC >12 000 μl−1; leucopaenia—WBC <4000 μl−1; hyperglycaemia (plasma glucose >7.7 mmol litre-1) in the absence of diabetes; plasma C-reactive protein >2 sd above the normal value |
| Sepsis | The clinical syndrome defined by the presence of both signs and symptoms of infection as described above and documented or suspected new infection |
| Severe sepsis | Sepsis complicated by one organ dysfunction |
| Multi-organ failure | Two or more organ dysfunction |
| Organ dysfunction | Defined using the definitions used for the Sequential Organ Failure Assessment (SOFA) score:40Pao2/Fio2≤26.7 kPa; noradrenaline/adrenaline ≥0.1 μg kg−1 min−1; creatinine ≥330 μmol litre−1; platelets ≤50×103 ml−1; bilirubin ≥102 μmol litre−1; Glasgow Coma Score <9 |
| Septic shock | In adults applies to a state of acute circulatory failure unexplained by other causes, defined as persistent arterial hypotension [a systolic arterial pressure (SBP) <90, MAP<60, or a reduction in SBP 40 mm Hg from baseline despite adequate volume resuscitation] |
Severe sepsis is common, the prevalence being approximately 2.3 cases per 100 hospital discharges in the USA, which translates into an annual burden of approximately 751 000 cases, and 68% of them require intensive care unit (ICU) treatment.12 The prevalence of severe sepsis in the first 24 h was estimated to be 27.1% in adult ICU in England, Wales, and Northern Ireland.93 Severe sepsis is frequently fatal, hospital mortality rate remains between 30% and 50%, or 500 000 deaths per year (1400 each day) worldwide, with as many deaths annually as those from acute myocardial infarction and the number is projected to grow at a rate of 1.5% per year.3
As the mortality rate of severe sepsis remains unacceptably poor, much research has been involved in the search for therapeutic interventions to improve outcomes. Over the last few years, several major clinical trials have finally shown that it is possible to reduce mortality in patients with sepsis.13,16,104,119 However, translating these recent therapeutic advances to routine clinical practice has proven controversial.15,82 Therapeutic interventions for management of severe sepsis and septic shock present a significant clinical challenge and new approaches and additional strategies continue to be developed. This review aims to assess the evidence of whether future treatment of sepsis would benefit from including statin therapy.
Pathophysiology of sepsis
Opinions on the causes and potential therapies for sepsis have evolved significantly over the last 20 years. Mounting evidence suggests that beside well-established factors, such as virulence of pathogens or site of infection, individual differences in disease manifestation are a result of the genetic predisposition of the patient with sepsis.126
In this review, we have concentrated upon those aspects of the basic mechanisms of the septic process that are particularly relevant to statin therapy.
Cytokine cascade: too much of a good thing?
The inflammatory response is a central component of sepsis as it drives the physiological alterations that are recognized as the SIRS.100 A successful inflammatory response eliminates the invading microorganisms without causing lasting damage. Sepsis develops when the initial, appropriate host response to an infection becomes amplified, and then aberrant. Bacterial components reacting with specific toll receptors are believed to trigger monocytes, neutrophils, and endothelial cells (EC) to initiate an inflammatory cascade70 (Fig. 1).
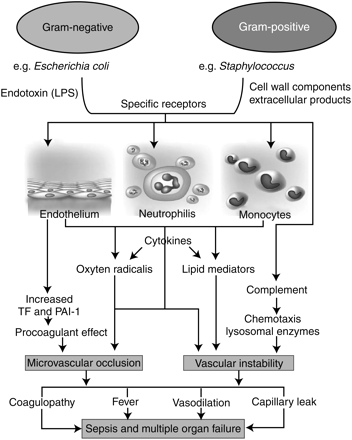
Pathophysiology of sepsis and multi-organ failure25 (adapted from Cohen J, Nature 2002, with permission).
Many believe that sepsis develops as a result of exuberant production of proinflammatory molecules such as TNF-α and IL-1, IL-6, and IL-8, lysosomal enzymes, superoxide-derived free radicals, vasoactive substances, such as platelet-activating factor (PAF), tissue factor (TF), and plasminogen activator inhibitor-1 (PAI-1).10 This occurs in conjunction with increases in the expression of inducible nitric oxide (NO) synthase, increasing production of NO resulting in coagulopathy, endothelial dysfunction, vascular instability, and eventually to apoptosis (i.e. programmed cell death) and multi-organ failure. Unfortunately, despite clinical trials of agents blocking single mediators, for example, TNF-α and IL-1 results have been disappointing. However, a meta-analysis of studies of all TNF inhibitors did demonstrate overall improvement.77
Given the complexity of sepsis syndrome, merely blocking a single component may be insufficient to arrest the inflammatory process.43 Consideration needs to be given to modulation of multiple targets which are central to the pathophysiological response in sepsis. Where activation of a critical part of the inflammatory pathway exhibits multiple or redundant pathways, we may need to intervene at two or more drivers of the process.107 Therefore, future strategies of intervention which modify several arms of the inflammatory cascade may possibly be more successful.82
Cytokine cascade: too little of a good thing?
In contrast to the hypothesis of exuberant inflammatory response in sepsis, is the finding that septic patients may have a relative anti-inflammatory environment. Thus, TNF:IL-10 ratios may be reduced, producing the equivalent of a blunted inflammatory response.117 Defective mediator production in response to stimuli has been seen in both monocytes and T lymphocytes.30 The blunted monocyte response seen in sepsis has been successfully reversed with interferon gamma and this proved effective in a small series of sepsis patients. However, a larger trial of trauma patients showed that although interferon gamma did reduce deaths due to infections, it did not reduce overall mortality.36
Vascular damage in sepsis
Recent attempts at a unifying hypothesis have been developed to explain the vascular changes in septic shock37 on the basis of the effect of inflammatory mediators on the vascular endothelium.28,57 The endothelium is involved in the control of vascular tone, vascular permeability, and coagulation,24 and a number of changes in it follow exposure of EC to relevant proinflammatory mediators.44 It has therefore been proposed that widespread endothelial damage and death occurs during human sepsis and leads to organ dysfunction and failure: the multi-organ dysfunction syndrome (MODS). This pan-endothelial damage is particularly important within the pulmonary circulation and results in high permeability pulmonary oedema that is clinically recognized as ARDS and necessitates mechanical ventilation.115
A role for endothelial apoptosis in sepsis?
Cellular death may be a key factor in sepsis and its related mortality. Cells that are destined to die can do so by two mechanisms: apoptosis and necrosis. The role of endothelial cell apoptosis in sepsis remains inconclusive due to the challenges involved in study of endothelial cell death in vivo. A major problem is that dying EC detach from the basement membrane and leave the vessel wall. Thus, apoptotic EC may not be detected on routine histology.50 Nevertheless, in humans soluble Fas levels are elevated in patients with multi-organ failure and levels fall during recovery.35,78 In addition, circulating levels of nuclear matrix protein, a general cell death index, is elevated in sepsis-related MODS patients and correlates with severity.127 Finally, circulating EC have been detected in the blood of patients with sepsis supporting a role of endothelial cell death in early sepsis.88
In sepsis, cytokine-induced coagulopathy triggers increased activity of TF and PAI-1 and decreased levels of the natural anticoagulant protein C on mononuclear and EC (Fig. 2).25 TF in turn activates a series of proteolytic cascades, which result in the conversion of prothrombin to thrombin, which in turn generates fibrin from fibrinogen. Simultaneously, PAI-1 prevents the conversion of plasminogen to plasmin, which results in impaired fibrinolysis. The activated form of protein C, aPC, formed when thrombin links with thrombomodulin (TM), inactivates factors Va and VIIa, and inhibits PAI-1 activity; hence, protein C deficiency results in further procoagulant effect. The net result is enhanced formation of fibrin clots in the microvasculature, leading to microvascular occlusion, impaired tissue perfusion, and then multi-organ failure.25
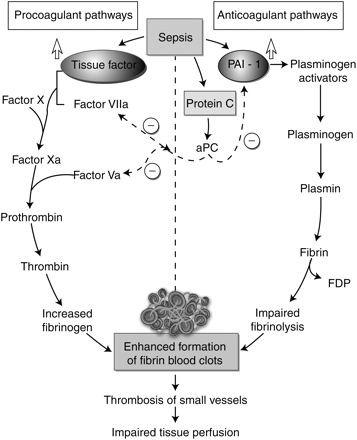
Mechanisms of coagulopathy in sepsis25 (adapted from Cohen J, Nature 2002, with permission).
Recombinant activated protein C (drotrecogin alfa, rhAPC) is currently the only pharmacological intervention available with anti-thrombotic and profibrinolytic property to treat severe sepsis. But rhAPC is expensive, can only be administered i.v., and bleeding is a risk.16 In addition, efficacy of activated protein C has been called into question recently and further research is being done to confirm the previous findings.
The need for new treatments in sepsis
Despite a considerable amount of research into the mechanisms of sepsis and investment in clinical trials of drugs that block single mediators, with the possible exception of activated protein C, no therapy targeting the inflammatory cascade has been shown to alter mortality in sepsis. Clearly, this represents a major healthcare need. The ideal agent would be cheap, have a clearly established and minimal side-effect profile, multiple modes of delivery, and have a pleiotropic effect upon the inflammatory cascade. We have assessed how close statins come to this ideal.
Pharmacology of statins
The statins are a class of lipid-lowering drugs which inhibit the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (Fig. 3). HMG-CoA reductase catalyses the conversion of hydroxymethylglutaryl-CoA to mevalonate, an early rate-limiting step in cholesterol biosynthesis, leading to cholesterol-lowering effects. In clinical studies, statins reduce total cholesterol, low-density lipid (LDL) cholesterol, apolipoprotein B, and triglyceride levels.
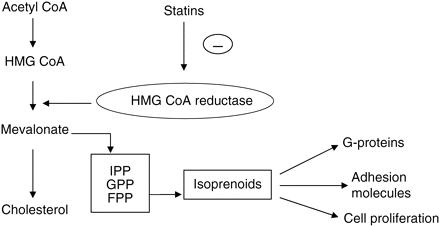
Mechanisms of hypolipidaemic effects by statins.
Seven drugs in the ‘statin’ class are now approved for clinical use in their hypolipidaemic effects in at least one country: lovastatin, pravastatin sodium, and simvastatin are fungal-derived inhibitors of HMG-CoA, whereas atorvastatin calcium, fluvastatin sodium, rosuvastatin, and pitavastatin are fully synthetic compounds.29 Cerivastatin sodium was withdrawn from the world market by the manufacturer in 2001 because of reports of fatal rhabdomyolysis.40 Six statins (not cerivastatin and pitavastatin) are currently used in the UK,1,17 although simvastatin and atorvastatin account for around 85% of all statin prescriptions in the UK.86
Rationale for statin use in sepsis: cell-based studies
Over the preceding decade, HMG-CoA reductase inhibitors have emerged as potentially powerful inhibitors of the inflammatory process44 (Table 2). The mechanism by which statins modulate the immune response is complex, but is often regarded as lipid independent as they are not related to lowering LDL cholesterol. Nevertheless, these effects primarily involve the inhibition of isoprenoid lipid production and subsequent protein prenylation and activation of signalling proteins such as the small GTPases.45
| Drugs | Solubility | Daily dose (mg) | Bioavailability (%) | Protein binding (%) | Metabolism | Elimination half-life (h) | Renal excretion (%) |
|---|---|---|---|---|---|---|---|
| Lovastatin | Lipophilic | 20–80 | 5 | >95 | CYP3A4 | 2–3 | 10 |
| Simvastatin | Lipophilic | 10–80 | 5 | 95–98 | CYP3A4 | 1–3 | 13 |
| Pravastatin | Hydrophilic | 10–40 | 20 | 43–67 | Sulfation | 2–3 | 20 |
| Flucastatin | Lipophilic | 20–80 | 24 | 98 | CYP2C9 | 0.5–3.0 | 6 |
| Atorvastatin | Lipophilic | 10–80 | 12 | 98 | CYP3A4 | 13–16 | 2 |
| Rosuvastatin | Hydrophilic | 10–40 | 20 | 90 | CYP2C9 | 19 | 10 |
| Pitavastatin | Lipophilic | 1–8 | 80 | 96 | Hepatic uptake | 11 | NA |
| Cerivastatin* | Lipophilic | 0.2–0.3 | 60 | 99 | CYP3A4 | 2–3 | 30 |
| Drugs | Solubility | Daily dose (mg) | Bioavailability (%) | Protein binding (%) | Metabolism | Elimination half-life (h) | Renal excretion (%) |
|---|---|---|---|---|---|---|---|
| Lovastatin | Lipophilic | 20–80 | 5 | >95 | CYP3A4 | 2–3 | 10 |
| Simvastatin | Lipophilic | 10–80 | 5 | 95–98 | CYP3A4 | 1–3 | 13 |
| Pravastatin | Hydrophilic | 10–40 | 20 | 43–67 | Sulfation | 2–3 | 20 |
| Flucastatin | Lipophilic | 20–80 | 24 | 98 | CYP2C9 | 0.5–3.0 | 6 |
| Atorvastatin | Lipophilic | 10–80 | 12 | 98 | CYP3A4 | 13–16 | 2 |
| Rosuvastatin | Hydrophilic | 10–40 | 20 | 90 | CYP2C9 | 19 | 10 |
| Pitavastatin | Lipophilic | 1–8 | 80 | 96 | Hepatic uptake | 11 | NA |
| Cerivastatin* | Lipophilic | 0.2–0.3 | 60 | 99 | CYP3A4 | 2–3 | 30 |
| Drugs | Solubility | Daily dose (mg) | Bioavailability (%) | Protein binding (%) | Metabolism | Elimination half-life (h) | Renal excretion (%) |
|---|---|---|---|---|---|---|---|
| Lovastatin | Lipophilic | 20–80 | 5 | >95 | CYP3A4 | 2–3 | 10 |
| Simvastatin | Lipophilic | 10–80 | 5 | 95–98 | CYP3A4 | 1–3 | 13 |
| Pravastatin | Hydrophilic | 10–40 | 20 | 43–67 | Sulfation | 2–3 | 20 |
| Flucastatin | Lipophilic | 20–80 | 24 | 98 | CYP2C9 | 0.5–3.0 | 6 |
| Atorvastatin | Lipophilic | 10–80 | 12 | 98 | CYP3A4 | 13–16 | 2 |
| Rosuvastatin | Hydrophilic | 10–40 | 20 | 90 | CYP2C9 | 19 | 10 |
| Pitavastatin | Lipophilic | 1–8 | 80 | 96 | Hepatic uptake | 11 | NA |
| Cerivastatin* | Lipophilic | 0.2–0.3 | 60 | 99 | CYP3A4 | 2–3 | 30 |
| Drugs | Solubility | Daily dose (mg) | Bioavailability (%) | Protein binding (%) | Metabolism | Elimination half-life (h) | Renal excretion (%) |
|---|---|---|---|---|---|---|---|
| Lovastatin | Lipophilic | 20–80 | 5 | >95 | CYP3A4 | 2–3 | 10 |
| Simvastatin | Lipophilic | 10–80 | 5 | 95–98 | CYP3A4 | 1–3 | 13 |
| Pravastatin | Hydrophilic | 10–40 | 20 | 43–67 | Sulfation | 2–3 | 20 |
| Flucastatin | Lipophilic | 20–80 | 24 | 98 | CYP2C9 | 0.5–3.0 | 6 |
| Atorvastatin | Lipophilic | 10–80 | 12 | 98 | CYP3A4 | 13–16 | 2 |
| Rosuvastatin | Hydrophilic | 10–40 | 20 | 90 | CYP2C9 | 19 | 10 |
| Pitavastatin | Lipophilic | 1–8 | 80 | 96 | Hepatic uptake | 11 | NA |
| Cerivastatin* | Lipophilic | 0.2–0.3 | 60 | 99 | CYP3A4 | 2–3 | 30 |
Statins have also been shown to directly bind to the leucocyte integrin LFA-1 (also known as CD11a/CD18) and so interfere with its principal ligand ICAM-1.125 However, since most of the effects of statins can be reversed with mevalonate supplementation, it is likely that non-HMG-CoA reductase effects are not a major mechanism. The cell processes affected by statins are ubiquitous within humans and therefore affect the function of most cell types. Thus, their impact on inflammation involves modulation of leucocyte biology, altered antigen presenting cell function, and changes in epithelial and EC. It is these pleiotropic actions of statins that underlie their potential effectiveness as immunomodulators in sepsis.
Effect of statins on the leucocyte response in sepsis
Sepsis results in a significant and time-dependent increase in leucocyte recruitment, adherence, and transmigration of leucocytes and P-selectin expression on vascular endothelium. Statins greatly reduce both lipopolysaccharide (LPS)-induced and Staphylococcus aureus α-toxin-induced leucocytes migration and leucocytes recruitment.33,99,105 Statins result in a significant reduction of leucocytes adhesion to endothelium by down-regulation of surface expression of endothelial cell adhesion molecule P-selectin, CD11b, and CD18 and by inhibition of lymphocyte function antigen-1 (LFA-1)-mediated leucocyte adhesion.99,124,129,131
Statins also affect monocyte functions. A double-blind, placebo-controlled study,89 randomized healthy volunteers to receive either simvastatin (80 mg day−1) or placebo for 4 days before i.v. LPS administration (20 IU kg−1), showed that simvastatin attenuated up-regulation of toll-like receptors (TLR) 4 and 2 on the surface of monocytes by more than half after LPS challenge (P<0.02). Suppressed TLR4 and TLR2 expression was associated with decreased circulating concentrations of TNF-α and monocyte chemoattractant protein-1.
Effect of statins on the drivers of sepsis
Statins affect the production of many acute phase reactants, such as IL-6, IL-8, TNF-α, monocyte chemoattractant protein-1 (MCP–1), and C-reactive protein (CRP).4,14,33,41,46,55,59,87,101–103,105,114 CRP is mainly produced by hepatocytes in response to IL-6. In an in vitro study,14 human hepatocytes were stimulated with IL-6 in the presence or absence of simvastatin and atorvastatin. Hepatocytes treated with statins showed significant inhibition of IL-6-induced CRP production. The reduction of CRP levels was more pronounced with atorvastatin than with other statins.14,58
Effect of statins on coagulation cascade
Statins improve platelet function and diminish procoagulant activity by a reduction in platelet aggregation, TF activity, reduced conversion of prothrombin to thrombin with resultant reduced thrombin activity, and a decline in levels of fibrinogen.5,26,49,52,62,79,90,112 Statins also seem to stimulate fibrinolysis by altering the level and activity of tissue plasminogen activator (tPA) and PAI-1.18,32,60In vitro, atorvastatin strongly increased the expression and functional activity of TM in human umbilical vein EC and human coronary artery EC,110 indicating a potential role in the treatment of protein C deficiency. This raises the possibility that statins, particularly atorvastatin, could offer a potential pharmacological treatment for severe sepsis.
Effect of statins on endothelial cell function
As discussed above, the microvascular circulation and particularly endothelial cell dysfunction is central to the pathogenesis of sepsis, and this was reviewed in the British Journal of Anaesthesia last year.81 As sepsis develops, EC become activated, participating in the inflammatory process and generating multiple inflammatory mediators and up-regulating adhesion molecule expression. Endothelial cell adhesion molecules not only act as docking structures for leucocytes, but also activate the signalling cascades required for successful leucocyte transmigration. The weight of evidence suggests that statins inhibit this process.
A reduction in the activation of pro-inflammatory transcription factors in EC also represents a key mechanism by which statins exert their immunomodulatory effects. This appears to be due to reduced levels of NF-kappaB and so statins can block the effects of cytokines such as TNF-α upon EC. These effects of statins on vascular endothelium have been closely associated with their ability to up-regulate endothelial NO synthase (eNOS) activity and enhance NO production.
Physiologically, eNOS is activated by the protein kinase Akt to produce endothelium-derived NO for controlling vasomotor activity.73 Sepsis results in a decrease in eNOS function and an increase in inducible NO synthase (iNOS) expression, contributing to the hypotension and resistance to vasopressor drugs that occur in septic shock.7,42,63,98 Statins increase eNOS activity by inducing Akt-mediated phosphorylation of eNOS,61 up-regulating eNOS expression,64–69 and activating eNOS directly.56 This may be anti-inflammatory locally on EC as NO prevents leucocyte chemotaxis and reduces endothelial adhesion molecule expression, which in turn attenuates leucocyte migration. Activation of the Akt pathway by statins may also be important as it leads to reduced endothelial cell apoptosis in vitro.
Statins have also been found to increase the number of circulating endothelial progenitor cells (EPCs).72 Indeed, statins induce angiogenesis by promoting the proliferation, migration, and survival of circulating EPC.122
Rationale for statin use in sepsis: animal studies
Clearly, statins affect many pathways in vitro that are believed to be involved in the pathogenesis of sepsis. These pleiotropic actions, therefore, provide a rationale for their use in inflammatory diseases such as sepsis. However, the in vitro studies alone do not provide a clear rationale for clinical studies in humans. It is for this reason that consideration of animal studies is important.
Several animal models have been used to look at the effects of statin therapy in sepsis. In a LPS-induced sepsis murine model, pre-treatment with cerivastatin significantly improved 7-day survival. Pre-treatment with cerivastatin also reduced serum levels of TNF-α and IL-1beta at 2 h and NO, nitrite, and nitrate at 8 h.9
In the mouse cecal ligation and puncture (CLP) model of polymicrobial sepsis, pre-treatment with simvastatin significantly increased both the rate of 3-day survival from 26% to 73%128 and duration of survival from 28 to 108 h85 compared with untreated mice. This improvement was associated with a preservation of cardiac function and haemodynamic status, which were severely impaired in untreated CLP mice. As one of the underlying mechanisms, the authors found increased mononuclear cell adhesiveness in septic mice, an important contributor to sepsis pathophysiology, to be reversed by statin treatment.85
A subsequent CLP study in mice demonstrated that this effect was seen with simvastatin, atorvastatin, and pravastatin but not with fluvastatin. Taken together, these studies provide significant animal model evidence to support the immunomodulatory effect of statins in sepsis and suggest that this is a class effect of statins.84
Evidence for anti-inflammatory effects of statins in vivo in humans
A randomized controlled trial (RCT)74 demonstrated that patients with acute coronary syndrome treated with atorvastatin 40 mg day−1 had a rapid reduction in CRP (a mean of 4 days after initiation of treatment) compared with placebo. Atorvastatin 40 mg day−1 produced 32% more effect than equipotent doses of fluvastatin, lovastatin, provastatin, or simvastatin in reducing CRP levels in patients with coronary heart disease.
Systemic inflammatory response occurs frequently after coronary artery bypass surgery, and it is strongly correlated with the risk of postoperative morbidity and mortality. In a double-blinded, placebo-controlled, randomized study, atorvastatin (20 mg day−1) for 3 weeks before surgery significantly reduced IL-6 and IL-8 release and neutrophil adhesion to the venous endothelium in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass.20
In inflammatory conditions other than ischaemic heart disease, statins have been shown to improve disease activity. For example, the trial of atorvastatin (40 mg day−1) in 116 patients with active rheumatoid arthritis (TARA)80 demonstrated a significant improvement in clinical disease activity score. CRP and erythrocyte sedimentation rate declined by 50% and 28%, respectively, in the atorvastatin group compared with the placebo group. These studies, therefore, indicate that statins are effective in decreasing systemic and vascular inflammation in humans in vivo.
Rationale for statin use in sepsis: clinical studies
The clinical studies that have described effects of statins in sepsis have either addressed the effects of statins in reducing sepsis incidence/severity or retrospectively looked at mortality in those taking statins who developed sepsis. Many of these studies have been elegantly reviewed by Chua and colleagues.21 We will concentrate upon the most recent studies (Table 3).
Recent studies outlining benefit for statin use in sepsis
| Author | Type of study | Setting | Outcome for statin group |
|---|---|---|---|
| van de Garde118 | Retrospective case–control | UK General Practice Research Database | OR 0.51 for pneumonia |
| Almog and colleagues6 | Prospective observational | Atherosclerosis patients from tertiary centre | RR 0.22 for death from infection |
| Gupta and colleagues47 | Prospective observational | Dialysis patients from national registry | IRR 0.41 for hospital admission for sepsis |
| Donnino and colleagues34 | Prospective observational | Emergency Department attendees from tertiary centre | OR 0.44 for death from infection |
| Frost and colleagues39 | Prospective observational case–control | Combined HMO databases | OR 0.49 for fatal pneumonia |
| Schlienger and colleagues109 | Retrospective case–control | UK National General Practice Research Database | OR 0.47 for fatal pneumonia |
| Author | Type of study | Setting | Outcome for statin group |
|---|---|---|---|
| van de Garde118 | Retrospective case–control | UK General Practice Research Database | OR 0.51 for pneumonia |
| Almog and colleagues6 | Prospective observational | Atherosclerosis patients from tertiary centre | RR 0.22 for death from infection |
| Gupta and colleagues47 | Prospective observational | Dialysis patients from national registry | IRR 0.41 for hospital admission for sepsis |
| Donnino and colleagues34 | Prospective observational | Emergency Department attendees from tertiary centre | OR 0.44 for death from infection |
| Frost and colleagues39 | Prospective observational case–control | Combined HMO databases | OR 0.49 for fatal pneumonia |
| Schlienger and colleagues109 | Retrospective case–control | UK National General Practice Research Database | OR 0.47 for fatal pneumonia |
Recent studies outlining benefit for statin use in sepsis
| Author | Type of study | Setting | Outcome for statin group |
|---|---|---|---|
| van de Garde118 | Retrospective case–control | UK General Practice Research Database | OR 0.51 for pneumonia |
| Almog and colleagues6 | Prospective observational | Atherosclerosis patients from tertiary centre | RR 0.22 for death from infection |
| Gupta and colleagues47 | Prospective observational | Dialysis patients from national registry | IRR 0.41 for hospital admission for sepsis |
| Donnino and colleagues34 | Prospective observational | Emergency Department attendees from tertiary centre | OR 0.44 for death from infection |
| Frost and colleagues39 | Prospective observational case–control | Combined HMO databases | OR 0.49 for fatal pneumonia |
| Schlienger and colleagues109 | Retrospective case–control | UK National General Practice Research Database | OR 0.47 for fatal pneumonia |
| Author | Type of study | Setting | Outcome for statin group |
|---|---|---|---|
| van de Garde118 | Retrospective case–control | UK General Practice Research Database | OR 0.51 for pneumonia |
| Almog and colleagues6 | Prospective observational | Atherosclerosis patients from tertiary centre | RR 0.22 for death from infection |
| Gupta and colleagues47 | Prospective observational | Dialysis patients from national registry | IRR 0.41 for hospital admission for sepsis |
| Donnino and colleagues34 | Prospective observational | Emergency Department attendees from tertiary centre | OR 0.44 for death from infection |
| Frost and colleagues39 | Prospective observational case–control | Combined HMO databases | OR 0.49 for fatal pneumonia |
| Schlienger and colleagues109 | Retrospective case–control | UK National General Practice Research Database | OR 0.47 for fatal pneumonia |
Does statin treatment reduce the incidence of infection in patients at risk of sepsis?
An examination of the UK General Practice Research Database to identify the risk of pneumonia in diabetic patients in relation to statin therapy118 found 4719 patients who developed pneumonia after starting statin therapy, and matched them with controls in a 1:3 ratio. Statin therapy reduced the risk. This effect persisted after adjustment for confounding and in sub-group analyses. A prospective observational study of 11 490 patients6 with atherosclerosis to assess death from infection, in which 50% of the patients were on statin therapy, found a mortality from infection of 0.9% in the statin patients compared with 4.1% in those not on statins. The relative risk of death from infection in statin patients was 0.22 (95% CI 0.17–0.28). Statin therapy remained beneficial after adjustment for confounding factors and propensity matching. A prospective study of the risk of hospital admission for sepsis in 1041 patients with chronic renal failure requiring dialysis47 found that 303 patients were hospitalized for sepsis. The risk of hospital admission for sepsis in statin users was 0.41 (95% CI 0.25–0.68), which was reduced further after adjustment for potential confounders and propensity matching for statin therapy. Similarly, a study using a large database combining data from several Healthcare Management Organizations39 examined the effect of statin therapy on death from influenza or COPD. Moderate dose statin therapy is defined as a dose ≥4 mg day−1 protected against death from pneumonia with an odds ratio of 0.49 (95% CI 0.26–0.76). In contrast, a recent study found that statins are not associated with reduced mortality or need for admission to an ICU in patients with pneumonia and reports of benefit in sepsis may be a result of confounding variables.75
Does statin therapy reduce the risk of developing severe sepsis?
In a prospective observational cohort study of 361 consecutive patients admitted with suspected or documented acute bacterial infection, severe sepsis developed in 19% of patients in the no pre-statin group compared with only 2.4% of the pre-statin group.8 More recently, a study48 using propensity-based matching, which accounted for each individual's likelihood of receiving a statin, yielded a cohort of 69 168 patients with cardiovascular disease, of whom half (n=34 584) received a statin and half (n=34 584) did not. They found that patients receiving statins had a 19–25% lower incidence of sepsis, severe sepsis, or fatal sepsis than those in controls [hazard ratio, HR (95% CI) 0.81 (0.72–0.91), 0.83 (0.70–0.97), and 0.75 (0.61–0.93), respectively].
Does statin therapy reduce mortality in sepsis?
A prospective observational study of 2036 patients with suspected infection attending the emergency department34 found that the 412 patients on statin therapy had a clinically and statistically significant reduction in mortality. Absolute mortality was 1.9% (95% CI 0.6–3.3%) in patients on statins when compared with 4.4% (95% CI 3.4–5.4%) in those not on statins. Multiple logistic regression was used to control for severity of illness and co-morbidities, and the odds ratio of death for statin patients was 0.44 (0.20–0.94, P=0.03). Schlienger and colleagues109 used the United Kingdom General Practice Research Database to identify 55 118 patients on statin therapy, and then match a subset of 1253 of these patients who developed pneumonia to controls in a 1:4 ratio. After adjustment for co-morbidity and frequency of visits to their physician, the authors found a large reduction in risk of fatal pneumonia with an odds ratio of 0.47 (95% CI 0.25–0.88).
Clinical studies summary
In summary, there is circumstantial evidence from retrospective database enquiries and observational studies that statins may be helpful in sepsis. However, these studies do not explain why the incidence of sepsis is increasing, despite raising the use of statins in the population and all of these studies have methodological flaws that mean they are poor surrogates for RCT.
Limitations of statins as a treatment for sepsis: side-effects
The American College of Cardiology/American Heart Association/National Heart, Lung, and Blood Institute has issued a clinical advisory on the use and safety of statins.95 Statins have proven to be extremely safe in the vast majority of patients receiving them. Few significant side-effects were observed in clinical trials, and post-marketing reports of adverse events have been very limited when considered in comparison with the very large number receiving these drugs.95 Two common side-effects are, however, particularly relevant in sepsis: liver dysfunction and myositis. Many changes in liver function occur during the course of sepsis. At the onset of sepsis, the liver increases most of its functional capacities. However, within a few days, detoxification processes have been shown to deteriorate as a first sign of septic liver failure.92 The cytochrome P450 system is of particular interest in the context of statin therapy in sepsis since the cytochrome P450 (CYP) isoform CYP3A4 serves as the major pathway for metabolism of lovastatin, simvastatin, atorvastatin, and cerivastatin.
Statins can adversely affect hepatic function. Elevated hepatic transaminases occur in 0.5–2.0% and are dose-dependent.19,51 Transaminase elevation is frequently reversed with a reduction in dose, or selection of another statin.53 Statins have not been shown to worsen the outcome in persons with chronic transaminase elevations due to hepatitis B or C, and treatment of hyperlipidaemia may actually improve transaminase elevations in individuals with fatty liver.11 Nevertheless, in the context of severe sepsis, where hepatic dysfunction is common, the use of statins may be contraindicated in some patients.
Abnormal creatinine kinase levels are also commonly encountered in sepsis patients, particularly those with invasive streptococcal disease.31 This represents a problem for statin therapy in sepsis since the most serious adverse effect associated with statin therapy is myopathy, which may progress to severe myositis and rhabdomyolysis.
Non-specific muscle aches or joint pains that are generally not associated with significant increases in creatine kinase (CK) are a common complaint in clinical trials.95 In placebo-controlled cholesterol lowering trials, the incidence of these complaints, generally reported as about 5%, is similar between placebo and active drug therapy, suggesting that they may not be drug-related.11,19,27,38,53,96 However, occasionally patients treated with a statin exhibit severe myositis characterized by muscle symptoms with increased CK levels. Failure to discontinue drug therapy can lead to rhabdomyolysis, myoglobinuria, and acute renal necrosis.76,97 It is not known whether patients with sepsis are more prone to the muscle side-effects of statins and this would require large clinical trials to answer.
Limitations of statins as a treatment for sepsis: mode of delivery
The current seven drugs in the ‘statin’ class are only available as an oral preparation. All statins are absorbed rapidly from gastrointestinal tract after administration, reaching peak plasma concentration within 4 h.23,94,116 The lack of an i.v. formulation or of a clear pharmacokinetic profile for statins in patients with sepsis would need to be addressed before their use could become widespread. As statins are highly protein bound, it is particularly important to define the influence of hypoalbuminaemia on statin bioavailability and toxicology.
The need for randomized trials of statins as an adjunctive therapy in sepsis
There is evidence from cell-based studies, animal models of sepsis, and observational clinical studies that statin treatment could be beneficial in patients with sepsis. However, not all clinical studies to date show a positive outcome. Clearly, there is need for properly conducted randomized placebo controlled trials in this context.83,91,113,123
There are several such trials presenting progress. The ASEPSIS trial intends to address the question as to whether statin therapy in early sepsis can modulate the development of severe sepsis compared with placebo. This trial is due to finish in 2008. The Intensive Care Society UK Clinical Trial group is investigating the effect of 40 mg simvastatin or placebo in patients with severe sepsis on plasma concentration of IL-6. In Belfast, the HARP study (ISRCTN70127774) is investigating simvastatin safety and effect on important surrogate clinical outcomes in 80 adult patients with sepsis-related acute lung injury. In Mexico, trial NCT00357123 is determining the effect of rosuvastatin on sepsis after abdominal surgery. Similarly, trial NCT00452608 is looking at the effect of atorvastatin upon inflammatory biomarkers in severe sepsis. In Vienna, trial NCT00450840 is looking at simvastatin and shock reversal, as defined by cessation of vasopressor support for more than 1 h. Unfortunately, all these trials are single centre studies that do not have mortality as the primary outcome variable.
Conclusions
The available evidence suggests that, in the future, treatment of sepsis may benefit from including statin therapy. Given their pleiotropic effects related to many pathophysiological determinants of sepsis, statin therapy may be the next logical step in the search for adjuvant therapy. However, there have been no RCT of statins in sepsis, and large randomized controlled clinical trials with clinically relevant primary endpoints are desperately needed. It is imperative to test the efficacy and safety and the benefit/adverse effects of statins administered at the onset of sepsis, as well as in patients with severe sepsis or septic shock admitted into ICUs.
References
Author notes
Declaration of interest. Professor Gao is the Chief Investigator of RCT on ‘Statin therapy in early sepsis can modulate the development of severe sepsis compared to placebo’ (ASEPSIS trial). The trial received a pump-prime research grant from Pfizer. Professor Gao and Dr Thickett have received travelling sponsorships from pharmaceutical and industrial companies to attend national and international conferences.



{kind=link}
{kind=link}
{kind=link}
Comments
We congratulate Gao et al (1) on the timely review of the place of statin therapy is sepsis. We agree that further prospective clinical research is required to evaluate the potential benefits and limitations of statin use in patients with sepsis and that such studies would need to specifically consider both current statin users and patients not taking statin therapy.
Stopping established statin therapy in patients with acute coronary disease (2), Recent major vascular surgery (3) or recent stroke (4) has been suggested to be associated with worse outcomes. This has not been specifically assessed in patients with sepsis, although a retrospective study by Kruger et al (5) in patients with bacteraemia showed continuing statin use after bacteremia was associated with significantly reduced mortality. These findings suggest that ceasing pre-existing statin therapy in sepsis (as recommended by current prescribing guidelines) may be associated with increased mortality. These findings require further evaluation in an appropriate prospective randomised trial.
Whilst the available evidence suggests that the potential for statins as adjuvant therapy in sepsis should be tested, we believe that an international multicentre trial with mortality as an endpoint would be premature. Preliminary data on absorption, pharmacokinetics, physiological effects and possible adverse effects in critically ill patients with sepsis are required.
With the support of the Australian and New Zealand Intensive Care (ANZIC) Clinical Trials Group and the ANZIC research Centre, we have commenced an Australian National Health and Medical Research Council (NHMRC)-funded multicentre phase II trial in 2007. The STATInS trial (ACTRN 12607000028404) (www.anzctr.org.au/trial_view.aspx?ID=81692) is a phase II, randomised, placebo-controlled study of the safety, pharmacokinetics and effect on inflammatory marker levels of atorvastatin in intensive care patients with severe sepsis. This trial is currently underway in more than 14 Intensive Care Units in Australia and New Zealand and we hope the results will provide a platform to plan future trials examining mortality as an end point.
The STATInS Trial investigators
References:
1 Gao F, Linhartova L, Johnston AM, et al. Statins and sepsis. Br J Anaesth 2008; 100:288-298
2 Heeschen C, Hamm CW, Laufs U, et al. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446-1452
3 Schouten O, Hoeks SE, Welten GM, et al. Effect of statin withdrawal on frequency of cardiac events after vascular surgery. Am J Cardiol 2007; 100:316-320
4 Blanco M, Nombela F, Castellanos M, et al. Statin treatment withdrawal in ischemic stroke: a controlled randomized study. Neurology 2007; 69:904-910
5 Kruger P, Fitzsimmons K, Cook D, et al. Statin therapy is associated with fewer deaths in patients with bacteraemia. Intensive Care Med 2006; 32:75-79
Conflict of Interest:
None declared