





Abstract
Exploring human-virus protein–protein interactions (PPIs) is crucial for unraveling the underlying pathogenic mechanisms of viruses. Limitations in the coverage and scalability of high-throughput approaches have impeded the identification of certain key interactions. Current popular computational methods adopt a two-stream pipeline to identify PPIs, which can only achieve relation modeling of protein pairs at the classification phase. However, the fitting capacity of the classifier is insufficient to comprehensively mine the complex interaction patterns between protein pairs.
In this study, we propose a pioneering single-stream framework HBFormer that combines hybrid attention mechanism and multimodal feature fusion strategy for identifying human-virus PPIs. The Transformer architecture based on hybrid attention can bridge the bidirectional information flows between human protein and viral protein, thus unifying joint feature learning and relation modeling of protein pairs. The experimental results demonstrate that HBFormer not only achieves superior performance on multiple human-virus PPI datasets but also outperforms 5 other state-of-the-art human-virus PPI identification methods. Moreover, ablation studies and scalability experiments further validate the effectiveness of our single-stream framework.
Codes and datasets are available at https://github.com/RmQ5v/HBFormer.
1 Introduction
Viruses are one of the major pathogenic agents of global infectious diseases. Within the intricate human-virus interaction system, protein–protein interactions (PPIs) are tightly linked with multiple pivotal biological processes underlying viral infection (Dyer et al. 2008, Lasso et al. 2019). The surface proteins of the virus can bind to specific cellular receptors in human, initiating viral invasion and triggering a series of intracellular signaling cascades, such as mediating cytoskeleton remodeling, activating protein kinases, and suppressing host cell immune response (Grove and Marsh 2011, Dey and Mondal 2024). Also due to the small genome, the virus has to exploit the established functions of human cellular proteins to complete its life cycle (Yang et al. 2019). Thus, identifying human-virus PPIs is critical for unraveling the underlying pathogenic mechanisms of viruses and for the development of targeted antiviral therapies.
Affinity purification coupled with mass spectrometry (AP-MS) and the yeast two-hybrid (Y2H) system have been extensively employed in enriching and identifying stable interaction partners of specific proteins (Brückner et al. 2009, Qin et al. 2021). Moreover, the Y2H system offers the advantage of capturing weak or transient protein interactions in actual cellular processes (Stynen et al. 2012). Such finely regulated processes exist for many important signaling proteins such as receptor kinases as well as drivers of cellular genealogy (Xing et al. 2014). However, even with substantial investments of time and resources in biological experiments, a great number of false positive results are inevitably generated and many known interactions are frequently missed (Peng et al. 2017). Consequently, computational approaches for identifying potential PPIs have been on the rise, providing testable hypotheses that act as a supplement for experimental efforts.
Numerous traditional machine learning approaches have made progress in the identification of PPIs (Yang et al. 2010, Cui et al. 2012, Dey et al. 2020). For instance, Dey et al. combined random forest with support vector machine to predict viral-host PPIs on protein datasets characterized by amino acid composition, pseudo amino acid composition, and conjoint triad. Yang et al. encoded protein sequence into vectors by local descriptors and leveraged k-nearest neighbor model to detect protein interaction patterns. However, these methods based on hand-crafted feature extraction have limited capabilities in constructing complex hierarchical feature representations, which restrict the classification performance of machine learning models. Given that feature extraction is the key to mining the information patterns of proteins, several tools that integrate protein feature extraction utilities have emerged, such as BioSeq-BLM (Li et al. 2021) and iLearnPlus (Chen et al. 2021).
In recent years, deep learning techniques have become a promising strategy for accelerating PPI identification research (Yang et al. 2022). DPPI (Hashemifar et al. 2018) was the first approach to deploy deep learning into identification of PPIs, which designed stacked convolutional neural modules to detect local patterns within sequence profiles, and then performed binary classification through linear layers. After that, PIPR (Chen et al. 2019) introduced a residual recurrent convolutional neural network (CNN) (LeCun and Bengio 1995) aimed at providing multi-granular sequence feature aggregation. TransPPI (Yang et al. 2021) mined and generated high-dimensional sequence embeddings from sequence profiles based on a Siamese CNN model to facilitate human-virus PPI identification. Nonetheless, dependencies of the global nature of protein sequences are difficult to capture by local convolution operations, and the lack of global features degrades the performance of deep learning approaches. To efficiently model long-distance dependencies of sequences, some works attempted to introduce long short-term memory (LSTM) (Hochreiter and Schmidhuber 1997) neural network. For instance, DNN-PPI (Li et al. 2018) constructed LSTM network to capture short-term dependencies at the amino acid level and long-term dependencies at the motif level. LSTM-PHV (Tsukiyama et al. 2021) leveraged LSTM with the word embedding model word2vec to learn the context information of amino acid sequences and derived predicted output via fully connected neural networks.
While the mentioned methods have achieved breakthroughs in PPI identification domain, they still suffer from certain limitations. On the one hand, they focus solely on sequence information of proteins, often overlooking other relevant factors acting on PPIs such as biological processes and molecular functions. For instance, post-translational modifications (PTMs) can alter the spatial conformation of proteins, thereby influencing the binding ability to interact with other proteins (Wang et al. 2022). On the other hand, existing approaches adopt a two-stream pipeline for PPI identification, which means that separate feature extraction (two-stream) is performed on each of the two input proteins, and then the two sets of feature vectors are concatenated and fed into a classifier to capture the relationships between proteins. However, for different datasets, the input proteins in the two-stream stage may originate from different species, posing a great challenge to the generalization ability of the feature extractor. Moreover, the two-stream framework is constrained to achieving the relation modeling of protein pairs exclusively at the classification phase. Common classifiers essentially aggregate multiple features in a certain proportion of weights, yet this aggregation capability is relatively limited. The fitting capacity of the classifier module is insufficient to comprehensively mine the complex interaction patterns between protein pairs.
To address the above challenges, we propose a single-stream framework named HBFormer, for identifying human-virus PPIs. This predictor is constructed based on hybrid attention mechanism and multimodal feature fusion strategy for proteins. The protein biological annotation features are tokenized to achieve interaction with sequence features in order to comprehensively characterize the intrinsic relationship between protein pairs, improving the prediction performance of the model. Meanwhile, the Transformer with hybrid attention can bridge the bidirectional information flows between human protein and viral protein, thereby unifies joint feature learning and relation modeling of protein pairs. Experimental results indicate that HBFormer outperforms other state-of-the-art methods and significantly impacts human-virus PPI identification.
2 Materials and methods
2.1 Overview of HBFormer
As shown in Fig. 1, the HBFormer framework encompasses three main parts: sequence embedding and feature extraction module, hybrid attention module based on multimodal features and interaction prediction module.
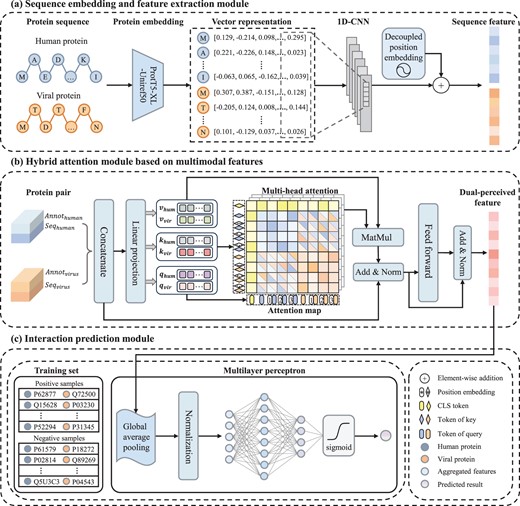
The framework of HBFormer. (a) Sequence embedding and feature extraction module. The pre-trained protein language model ProtT5-XL-Uniref50 and decoupled position encoding are applied to generate informative sequence feature representations of human proteins and viral proteins. (b) Hybrid attention module based on multimodal features. The protein biological annotation features are tokenized to achieve interaction with sequence features to comprehensively characterize the intrinsic relationship between protein pairs. The hybrid attention has dual attention operations, where self-attention allows to learn and extract features from protein itself while cross-attention aims to model the relationship between human protein and viral protein. The dual-perceived features for each of the protein pairs can be obtained in this module. (c) Interaction prediction module. A multilayer perceptron is employed to learn hidden representations from dual-perceived features and calculate predicted probability score of interaction.
2.2 Sequence feature representation
Pre-trained protein language models are a powerful paradigm for creating effective embeddings and learning context-aware data representations and have been successfully used in various downstream protein understanding tasks (Liu and Tian 2023, Wang et al. 2023). In this study, we adopt ProtT5-XL-Uniref50 (PT5) (Elnaggar et al. 2022) to embed human proteins and viral proteins for more informative sequence representations. PT5 is a pre-trained model based on the Text-to-Text Transfer Transformer (T5) (Raffel et al. 2020) architecture, conducted in a self-supervised manner through masked language modeling. This model is not only capable of capturing complex pattern in sequence contexts, but also generating different embedding representations of the same amino acid according to different contextual semantic information. It comprises a 24-layer encoder–decoder, encompassing 3B parameters. The pretext task of PT5 is to reconstruct inputs from masked tokens (amino acids), aiming at training a more powerful feature encoder. The whole process is trained on the BFD (Steinegger and Söding 2018) protein sequence corpora, which contain 393 billion amino acids, and then fine-tuned on the UniRef50 (Suzek et al. 2015) dataset. In implementation, we freeze the weights of the fine-tuned PT5 model to encode sequence. Each amino acid in the human and viral protein sequence is output as a 1024-dimensional embedding vector.
2.3 Biological annotation feature representation
Due to the large number of entries encompassed by the biological annotation data, the dimensionality of features is correspondingly high. Consequently, principal component analysis is introduced to reduce the dimensionality of the biological annotation features and to mitigate potential noise effects. We preserve 98% of the biological annotation feature information for both human proteins and viral proteins.
2.4 Transformer based on hybrid attention mechanism with multimodal features
2.5 Prediction by multi-layer perceptron
3 Results
3.1 Dataset
Benchmark dataset
To measure the performance of HBFormer for identifying human-virus PPIs, we adopt the high-quality benchmark dataset assembled by Tsukiyama et al. (2021). The positive samples contain experimentally validated 22 383 PPIs involving in 5882 human proteins and 996 viral proteins. Among the data, the confidence score of each interaction is greater than 0.3. Meanwhile, all proteins are non-redundant, composed of standard amino acids, and had lengths between 30 and 1000 residues. Regarding the construction of negative samples, the dissimilarity negative sampling method (Eid et al. 2016) is utilized to effectively mitigate the impact of noise samples that are similar to positive samples. The ratio of positive and negative samples in this benchmark dataset is 1:10. All samples are divided into a training set (conducting cross-validation) and an independent test set with a ratio of 8:2.
Specific types of human-virus PPI dataset
In order to comprehensively evaluate the performance of HBFormer, we use the dataset from the TransPPI work (Yang et al. 2021). The TransPPI dataset contains experimentally validated information on the interactions between human proteins and proteins of eight specific types of viruses, which are HIV, Herpes, Papilloma, Influenza, Hepatitis, Dengue, Zika, and SARS-CoV-2. The number of positive samples is 9880, 5966, 5099, 3044, 1300, 927, 709, and 568, respectively. Similarly, the negative samples of this dataset are obtained by the dissimilarity negative sampling method with a ratio of positive to negative samples of 1:10.
Non-viral pathogen PPI dataset
To investigate the applicability of our method to other types of pathogens, the interaction dataset is taken from Kösesoy et al. (2019), which includes PPIs between human and Bacillus anthracis, as well as human and Yersinia pestis. In the Human-Bacillus anthracis PPI dataset, the number of positive and negative interactions is 3090 and 9500, respectively. The Human-Yersinia pestis PPI dataset consists of 4097 positive samples and 12 500 negative samples. Negative samples for both datasets are generated using the random sampling method.
3.2 Comparison with state-of-the-art methods
To verify the effectiveness of our model, we compare HBFormer with five other state-of-the-art human-virus PPI identification methods on the benchmark dataset. As shown in Table 1, HBFormer based on the multi-feature fusion strategy surpasses other competing methods across all metrics. When HBFormer solely uses sequence features, it also exhibits superior performance in AUC and AUPRC metrics compared to other approaches, and the higher AUPRC value demonstrates the effectiveness of our single-stream framework in identifying positive examples. We can also observe that the AUPRC of HBFormer is 23.5% higher than that of the DeepViral method (Liu-Wei et al. 2021) with the multi-feature fusion strategy, as well as showing higher values in terms of MCC and F1 scores, which can provide better predictions when faced with an imbalance of data. Moreover, HBFormer significantly outperforms the sequence-based identification approaches (Yang et al. 2020, Tsukiyama et al. 2021, Yang et al. 2021, Madan et al. 2022), achieving improvements of 4.4%, 4.9%, 5.3%, and 18.2% on AUPRC, which may be attributed to the following key factors: On the one hand, we incorporate comprehensive protein characterization, encompassing not only sequences but also multifaceted biological annotation information, which provides the model with multidimensional insights into proteins. On the other hand, our pioneering single-stream framework is capable of performing additional and enhanced relation modeling between human protein and viral protein prior to classification, thus improving the model’s ability to discriminate interaction patterns.
Comparison of different methods on benchmark dataset.
| Method | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| DeepViral | 0.981 | 0.757 | 0.952 | 0.688 | 0.674 |
| Yang | 0.963 | 0.810 | 0.947 | 0.724 | 0.697 |
| LSTM-PHV | 0.976 | 0.939 | 0.984 | 0.910 | 0.903 |
| TransPPI | 0.982 | 0.943 | 0.984 | 0.896 | 0.886 |
| STEP | 0.983 | 0.948 | 0.983 | 0.908 | 0.902 |
| HBFormera | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| HBFormerb | 0.998 | 0.992 | 0.993 | 0.962 | 0.958 |
| Method | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| DeepViral | 0.981 | 0.757 | 0.952 | 0.688 | 0.674 |
| Yang | 0.963 | 0.810 | 0.947 | 0.724 | 0.697 |
| LSTM-PHV | 0.976 | 0.939 | 0.984 | 0.910 | 0.903 |
| TransPPI | 0.982 | 0.943 | 0.984 | 0.896 | 0.886 |
| STEP | 0.983 | 0.948 | 0.983 | 0.908 | 0.902 |
| HBFormera | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| HBFormerb | 0.998 | 0.992 | 0.993 | 0.962 | 0.958 |
Based on single-modal (sequence) features.
Based on multimodal (sequence+biological annotation) features.
Comparison of different methods on benchmark dataset.
| Method | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| DeepViral | 0.981 | 0.757 | 0.952 | 0.688 | 0.674 |
| Yang | 0.963 | 0.810 | 0.947 | 0.724 | 0.697 |
| LSTM-PHV | 0.976 | 0.939 | 0.984 | 0.910 | 0.903 |
| TransPPI | 0.982 | 0.943 | 0.984 | 0.896 | 0.886 |
| STEP | 0.983 | 0.948 | 0.983 | 0.908 | 0.902 |
| HBFormera | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| HBFormerb | 0.998 | 0.992 | 0.993 | 0.962 | 0.958 |
| Method | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| DeepViral | 0.981 | 0.757 | 0.952 | 0.688 | 0.674 |
| Yang | 0.963 | 0.810 | 0.947 | 0.724 | 0.697 |
| LSTM-PHV | 0.976 | 0.939 | 0.984 | 0.910 | 0.903 |
| TransPPI | 0.982 | 0.943 | 0.984 | 0.896 | 0.886 |
| STEP | 0.983 | 0.948 | 0.983 | 0.908 | 0.902 |
| HBFormera | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| HBFormerb | 0.998 | 0.992 | 0.993 | 0.962 | 0.958 |
Based on single-modal (sequence) features.
Based on multimodal (sequence+biological annotation) features.
3.3 Comparison across specific types of PPI datasets
To further assess the predictive ability of HBFormer across specific types of human-virus PPI datasets, we compare HBFormer with three other advanced identification methods that achieved high performance on the benchmark dataset. The results of the 5-fold cross-validation are shown in Table 2. It can be observed that the AUC metrics of our model show superiority over other advanced methods across all types of human-virus PPI datasets, especially on datasets with smaller data sizes (Hepatitis, Dengue, Zika virus, and SARS-CoV-2). Moreover, except for the slightly lower AUPRC on the Herpes and SARS-CoV-2 datasets compared with the STEP method (Madan et al. 2022), HBFormer achieves the highest AUPRC value on the other six datasets. These results imply that our single-stream model exhibits robust classification performance and reliability in uncovering interaction patterns, which is of great importance for advancing the identification of human-virus PPIs.
Comparison results across specific types of human-virus PPI dataset.
| Dataset | Method | AUC | AUPRC | ACC | Dataset | Method | AUC | AUPRC | ACC |
|---|---|---|---|---|---|---|---|---|---|
| Human-HIV | TransPPI | 0.995 | 0.974 | 0.986 | Human-Hepatitis | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.994 | 0.959 | 0.981 | LSTM-PHV | 0.908 | 0.559 | 0.920 | ||
| STEP | 0.996 | 0.976 | 0.988 | STEP | 0.901 | 0.639 | 0.926 | ||
| HBformer | 0.996 | 0.976 | 0.989 | HBformer | 0.935 | 0.685 | 0.940 | ||
| Human-Herpes | TransPPI | 0.941 | 0.768 | 0.952 | Human-Dengue | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.936 | 0.722 | 0.935 | LSTM-PHV | 0.891 | 0.457 | 0.905 | ||
| STEP | 0.951 | 0.802 | 0.956 | STEP | 0.924 | 0.638 | 0.933 | ||
| HBformer | 0.961 | 0.781 | 0.954 | HBformer | 0.958 | 0.683 | 0.940 | ||
| Human-Papilloma | TransPPI | 0.959 | 0.818 | 0.959 | Human-Zika | TransPPI | 0.926 | 0.746 | 0.954 |
| LSTM-PHV | 0.954 | 0.749 | 0.942 | LSTM-PHV | 0.867 | 0.571 | 0.924 | ||
| STEP | 0.962 | 0.823 | 0.957 | STEP | 0.937 | 0.757 | 0.949 | ||
| HBformer | 0.975 | 0.835 | 0.961 | HBformer | 0.951 | 0.802 | 0.953 | ||
| Human-Influenza | TransPPI | 0.964 | 0.834 | 0.961 | Human-SARS-CoV-2 | TransPPI | 0.805 | 0.329 | 0.906 |
| LSTM-PHV | 0.960 | 0.781 | 0.948 | LSTM-PHV | 0.776 | 0.253 | 0.869 | ||
| STEP | 0.959 | 0.843 | 0.958 | STEP | 0.837 | 0.428 | 0.911 | ||
| HBformer | 0.965 | 0.849 | 0.962 | HBformer | 0.879 | 0.424 | 0.895 |
| Dataset | Method | AUC | AUPRC | ACC | Dataset | Method | AUC | AUPRC | ACC |
|---|---|---|---|---|---|---|---|---|---|
| Human-HIV | TransPPI | 0.995 | 0.974 | 0.986 | Human-Hepatitis | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.994 | 0.959 | 0.981 | LSTM-PHV | 0.908 | 0.559 | 0.920 | ||
| STEP | 0.996 | 0.976 | 0.988 | STEP | 0.901 | 0.639 | 0.926 | ||
| HBformer | 0.996 | 0.976 | 0.989 | HBformer | 0.935 | 0.685 | 0.940 | ||
| Human-Herpes | TransPPI | 0.941 | 0.768 | 0.952 | Human-Dengue | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.936 | 0.722 | 0.935 | LSTM-PHV | 0.891 | 0.457 | 0.905 | ||
| STEP | 0.951 | 0.802 | 0.956 | STEP | 0.924 | 0.638 | 0.933 | ||
| HBformer | 0.961 | 0.781 | 0.954 | HBformer | 0.958 | 0.683 | 0.940 | ||
| Human-Papilloma | TransPPI | 0.959 | 0.818 | 0.959 | Human-Zika | TransPPI | 0.926 | 0.746 | 0.954 |
| LSTM-PHV | 0.954 | 0.749 | 0.942 | LSTM-PHV | 0.867 | 0.571 | 0.924 | ||
| STEP | 0.962 | 0.823 | 0.957 | STEP | 0.937 | 0.757 | 0.949 | ||
| HBformer | 0.975 | 0.835 | 0.961 | HBformer | 0.951 | 0.802 | 0.953 | ||
| Human-Influenza | TransPPI | 0.964 | 0.834 | 0.961 | Human-SARS-CoV-2 | TransPPI | 0.805 | 0.329 | 0.906 |
| LSTM-PHV | 0.960 | 0.781 | 0.948 | LSTM-PHV | 0.776 | 0.253 | 0.869 | ||
| STEP | 0.959 | 0.843 | 0.958 | STEP | 0.837 | 0.428 | 0.911 | ||
| HBformer | 0.965 | 0.849 | 0.962 | HBformer | 0.879 | 0.424 | 0.895 |
Comparison results across specific types of human-virus PPI dataset.
| Dataset | Method | AUC | AUPRC | ACC | Dataset | Method | AUC | AUPRC | ACC |
|---|---|---|---|---|---|---|---|---|---|
| Human-HIV | TransPPI | 0.995 | 0.974 | 0.986 | Human-Hepatitis | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.994 | 0.959 | 0.981 | LSTM-PHV | 0.908 | 0.559 | 0.920 | ||
| STEP | 0.996 | 0.976 | 0.988 | STEP | 0.901 | 0.639 | 0.926 | ||
| HBformer | 0.996 | 0.976 | 0.989 | HBformer | 0.935 | 0.685 | 0.940 | ||
| Human-Herpes | TransPPI | 0.941 | 0.768 | 0.952 | Human-Dengue | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.936 | 0.722 | 0.935 | LSTM-PHV | 0.891 | 0.457 | 0.905 | ||
| STEP | 0.951 | 0.802 | 0.956 | STEP | 0.924 | 0.638 | 0.933 | ||
| HBformer | 0.961 | 0.781 | 0.954 | HBformer | 0.958 | 0.683 | 0.940 | ||
| Human-Papilloma | TransPPI | 0.959 | 0.818 | 0.959 | Human-Zika | TransPPI | 0.926 | 0.746 | 0.954 |
| LSTM-PHV | 0.954 | 0.749 | 0.942 | LSTM-PHV | 0.867 | 0.571 | 0.924 | ||
| STEP | 0.962 | 0.823 | 0.957 | STEP | 0.937 | 0.757 | 0.949 | ||
| HBformer | 0.975 | 0.835 | 0.961 | HBformer | 0.951 | 0.802 | 0.953 | ||
| Human-Influenza | TransPPI | 0.964 | 0.834 | 0.961 | Human-SARS-CoV-2 | TransPPI | 0.805 | 0.329 | 0.906 |
| LSTM-PHV | 0.960 | 0.781 | 0.948 | LSTM-PHV | 0.776 | 0.253 | 0.869 | ||
| STEP | 0.959 | 0.843 | 0.958 | STEP | 0.837 | 0.428 | 0.911 | ||
| HBformer | 0.965 | 0.849 | 0.962 | HBformer | 0.879 | 0.424 | 0.895 |
| Dataset | Method | AUC | AUPRC | ACC | Dataset | Method | AUC | AUPRC | ACC |
|---|---|---|---|---|---|---|---|---|---|
| Human-HIV | TransPPI | 0.995 | 0.974 | 0.986 | Human-Hepatitis | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.994 | 0.959 | 0.981 | LSTM-PHV | 0.908 | 0.559 | 0.920 | ||
| STEP | 0.996 | 0.976 | 0.988 | STEP | 0.901 | 0.639 | 0.926 | ||
| HBformer | 0.996 | 0.976 | 0.989 | HBformer | 0.935 | 0.685 | 0.940 | ||
| Human-Herpes | TransPPI | 0.941 | 0.768 | 0.952 | Human-Dengue | TransPPI | 0.917 | 0.636 | 0.934 |
| LSTM-PHV | 0.936 | 0.722 | 0.935 | LSTM-PHV | 0.891 | 0.457 | 0.905 | ||
| STEP | 0.951 | 0.802 | 0.956 | STEP | 0.924 | 0.638 | 0.933 | ||
| HBformer | 0.961 | 0.781 | 0.954 | HBformer | 0.958 | 0.683 | 0.940 | ||
| Human-Papilloma | TransPPI | 0.959 | 0.818 | 0.959 | Human-Zika | TransPPI | 0.926 | 0.746 | 0.954 |
| LSTM-PHV | 0.954 | 0.749 | 0.942 | LSTM-PHV | 0.867 | 0.571 | 0.924 | ||
| STEP | 0.962 | 0.823 | 0.957 | STEP | 0.937 | 0.757 | 0.949 | ||
| HBformer | 0.975 | 0.835 | 0.961 | HBformer | 0.951 | 0.802 | 0.953 | ||
| Human-Influenza | TransPPI | 0.964 | 0.834 | 0.961 | Human-SARS-CoV-2 | TransPPI | 0.805 | 0.329 | 0.906 |
| LSTM-PHV | 0.960 | 0.781 | 0.948 | LSTM-PHV | 0.776 | 0.253 | 0.869 | ||
| STEP | 0.959 | 0.843 | 0.958 | STEP | 0.837 | 0.428 | 0.911 | ||
| HBformer | 0.965 | 0.849 | 0.962 | HBformer | 0.879 | 0.424 | 0.895 |
3.4 Comparison with popular embedding methods
To probe the capacity of different embedding methods for characterizing protein sequences, we train our model using four other widely used encoding schemes. Note that we construct HBFormer solely based on sequence features to ensure unbiased evaluation. For the static embedding methods Word2vec (Mikolov et al. 2013) and FastText (Joulin et al. 2016), we additionally fine-tune these models by using the genism library for a more comprehensive comparison. The results in Table 3 demonstrate that HBFormer with PT5 embedding exhibits improved performance compared with other encoding approaches. Specifically, without any fine-tuning on the benchmark dataset, PT5 shows a significant advantage compared with Word2vec and FastText in terms of the AUPRC. We also observe that these static embedding methods can lead to a significant improvement in the performance of classification models after fine-tuning on the dataset. However, their performance is still inferior to the PT5 method based on self-supervised training. This is attributed to PT5’s ability to learn context-specific sequence semantic information, enabling the generation of robust expressions enriched with more global contextual information and long-distance dependencies. In contrast to the pre-trained protein language model ESM-1B (Rives et al. 2021), which is also based on self-supervised training, PT5 presents superior performance across all metrics, indicating it can achieve more informative sequence representations. Furthermore, the AUC and AUPRC of PT5 are 2.9% and 12.7% higher than those of the method Bepler (Bepler and Berger 2019), which utilizes global structural similarity between proteins for weakly supervised embedding training, further illustrating the powerful expressive capabilities of sequence features derived from PT5.
Comparison results of different embedding methods.
| Methods | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Word2vec | 0.890 | 0.519 | 0.889 | 0.526 | 0.482 |
| Word2veca | 0.971 | 0.926 | 0.978 | 0.878 | 0.867 |
| FastText | 0.907 | 0.580 | 0.889 | 0.550 | 0.515 |
| FastTexta | 0.974 | 0.924 | 0.979 | 0.873 | 0.861 |
| ESM-1B | 0.949 | 0.761 | 0.924 | 0.656 | 0.639 |
| Bepler | 0.956 | 0.821 | 0.944 | 0.726 | 0.700 |
| PT5 | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| Methods | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Word2vec | 0.890 | 0.519 | 0.889 | 0.526 | 0.482 |
| Word2veca | 0.971 | 0.926 | 0.978 | 0.878 | 0.867 |
| FastText | 0.907 | 0.580 | 0.889 | 0.550 | 0.515 |
| FastTexta | 0.974 | 0.924 | 0.979 | 0.873 | 0.861 |
| ESM-1B | 0.949 | 0.761 | 0.924 | 0.656 | 0.639 |
| Bepler | 0.956 | 0.821 | 0.944 | 0.726 | 0.700 |
| PT5 | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
Fine-tuned on the benchmark dataset.
Comparison results of different embedding methods.
| Methods | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Word2vec | 0.890 | 0.519 | 0.889 | 0.526 | 0.482 |
| Word2veca | 0.971 | 0.926 | 0.978 | 0.878 | 0.867 |
| FastText | 0.907 | 0.580 | 0.889 | 0.550 | 0.515 |
| FastTexta | 0.974 | 0.924 | 0.979 | 0.873 | 0.861 |
| ESM-1B | 0.949 | 0.761 | 0.924 | 0.656 | 0.639 |
| Bepler | 0.956 | 0.821 | 0.944 | 0.726 | 0.700 |
| PT5 | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
| Methods | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Word2vec | 0.890 | 0.519 | 0.889 | 0.526 | 0.482 |
| Word2veca | 0.971 | 0.926 | 0.978 | 0.878 | 0.867 |
| FastText | 0.907 | 0.580 | 0.889 | 0.550 | 0.515 |
| FastTexta | 0.974 | 0.924 | 0.979 | 0.873 | 0.861 |
| ESM-1B | 0.949 | 0.761 | 0.924 | 0.656 | 0.639 |
| Bepler | 0.956 | 0.821 | 0.944 | 0.726 | 0.700 |
| PT5 | 0.985 | 0.948 | 0.984 | 0.907 | 0.898 |
Fine-tuned on the benchmark dataset.
3.5 Ablation study
To investigate the contribution of the hybrid attention mechanism, we test the performance of HBFormer with its self-attention module or cross-attention module removed. In this section, we train the HBFormer only using sequence features to eliminate potential biases introduced by biological annotation features. The results of the ablation study conducted on the independent test set are shown in Fig. 2a. It can be observed that the performance of HBFormer seems to decrease when we remove any part of the attention components. This elucidates that both self-attention and cross-attention contribute to the identification of interactions, but the impact on the model’s performance is greater in the absence of cross-attention, which may be due to the fact that cross-attention can provide additional relationship modeling and thus more effectively mine the relationships between proteins. Notably, HBFormer can be categorized as a two-stream framework when utilizing only self-attention, as in this stage, the Transformer only amounts to separate feature extraction operations for each of the two proteins. This further suggests that the single-stream attention mechanism framework outperforms the two-stream framework in the PPI identification tasks.
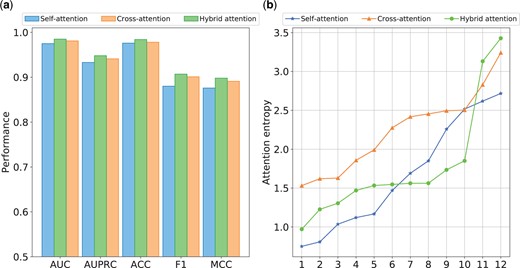
Ablation studies. (a) Performance comparison using different attention mechanism. (b) Attention entropy distribution across all attention heads using different attention mechanism.
In this study, we use a total of 12 heads based on multi-head attention mechanism. The average attention entropy distribution across all attention heads is shown in Fig. 2b. The HBFormer based on the hybrid attention mechanism exhibits a broader distribution of attention entropy compared with models employing only self-attention or cross-attention, which means that the attention heads have different specialization roles, allowing the model to better aggregate both local and global tokens with both concentrated and broad focuses. The above results prove that the hybrid attention mechanism is indispensable in our single-stream framework, facilitating the discrimination between interacting and non-interacting protein pairs.
3.6 Identification of non-viral pathogens PPIs
In order to further assess the generalizability of our proposed framework, we apply HBFormer to identify the interactions of human proteins with other non-viral pathogen proteins. We perform 5-fold cross-validation on the PPI datasets of Human-Bacillus anthracis (Human-B) and Humans-Yersinia pestis (Human-Y), respectively, and average the performance metrics of the subset models to produce more reliable predictions. The results are presented in Table 4. HBFormer still shows commendable performance on the two cross-species datasets, with AUC, AUPRC, and ACC all reaching above 0.9, suggesting that HBFormer can also be effectively extended to the PPI identification in humans and other types of pathogens.
Results on non-viral pathogens datasets.
| Dataset | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Human-B | 0.973 | 0.928 | 0.949 | 0.874 | 0.825 |
| Humans-Y | 0.961 | 0.909 | 0.935 | 0.853 | 0.792 |
| Dataset | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Human-B | 0.973 | 0.928 | 0.949 | 0.874 | 0.825 |
| Humans-Y | 0.961 | 0.909 | 0.935 | 0.853 | 0.792 |
Results on non-viral pathogens datasets.
| Dataset | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Human-B | 0.973 | 0.928 | 0.949 | 0.874 | 0.825 |
| Humans-Y | 0.961 | 0.909 | 0.935 | 0.853 | 0.792 |
| Dataset | AUC | AUPRC | ACC | F1 | MCC |
|---|---|---|---|---|---|
| Human-B | 0.973 | 0.928 | 0.949 | 0.874 | 0.825 |
| Humans-Y | 0.961 | 0.909 | 0.935 | 0.853 | 0.792 |
4 Discussion
In this paper, wek propose HBFormer, a single-stream computational framework based on the hybrid attention mechanism for identifying human-virus PPIs. Unlike other deep learning approaches for PPI identification, our pioneering single-stream framework is capable of achieving additional relation modeling between human protein and viral protein simultaneously with feature extraction, thereby improving the model’s ability to discriminate interaction patterns. Moreover, the multimodal feature fusion strategy adopted is also an important factor contributing to the excellent performance of HBFormer, which provides the model with both comprehensive and multidimensional insights into proteins. Extensive experiments validate the effectiveness of our proposed framework and demonstrate its extensibility to other types of PPI identification. However, there remain some shortcomings in this research. The imbalance learning strategy we incorporate may still not fully resolve the problem of skewed sample distributions in some categories, and thus more ways to rectify the imbalance still need to be explored in order to improve the performance of the model. Furthermore, due to the high computation and memory demands of the Transformer architecture, insufficient computational resources may affect the application of the model. Optimizing computation to break through speed bottlenecks with FlashAttention tools will be an extensible work in the future.
Conflict of interest: The authors declare no competing interests.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 62331012).
Data availability
All data and source code used in the present study is available at https://github.com/RmQ5v/HBFormer.


{kind=link}
{kind=link}