





Abstract
Drug repurposing is an approach used to discover new indications for existing drugs. Recently, several computational approaches have been developed for drug repurposing in cancer. Nevertheless, no approaches have reported a systematic analysis of pathway crosstalk. Pathway crosstalk, which refers to the phenomenon of interaction or cooperation between pathways, is a critical aspect of tumor pathways that allows cancer cells to survive and acquire resistance to drug therapy. Here, we innovatively developed a system biology R-based software package, DRviaSPCN, to repurpose drugs for cancer via a subpathway (SP) crosstalk network. This package provides a novel approach to prioritize cancer candidate drugs by considering drug-induced SPs and their crosstalk effects. The operation modes mainly include construction of the SP network and calculation of the centrality scores of SPs to reflect the influence of SP crosstalk, calculation of enrichment scores of drug- and disease-induced dysfunctional SPs and weighted them by the centrality scores of SPs, evaluation of the drug–disease reverse association at the weighted SP level, identification of cancer candidate drugs and visualization of the results. Its capabilities enable DRviaSPCN to find cancer candidate drugs, which will complement the recent tools which did not consider crosstalk among pathways/SPs. DRviaSPCN may help to facilitate the development of drug discovery.
The package is implemented in R and available under GPL-2 license from the CRAN website (https://CRAN.R-project.org/package=DRviaSPCN).
Supplementary data are available at Bioinformatics online.
Introduction
Drug repurposing in silico approaches has attracted wide attention from researchers due to its advantages such as low cost and high efficiency (Pushpakom et al., 2019). Recently, several computational approaches have been developed to identify cancer candidate drugs. For instance, drug verse disease provides a pipeline for drug repurposing through comparing drug and disease gene expression profiles (GEP) (Pacini et al., 2013), and Connectivity Map (CMap) prioritizes candidate drugs through evaluating the reverse association extent between gene expression induced by drugs and diseases (Lamb et al., 2006). Furthermore, in our previous studies, the SPs defined as the local gene subregions within a biological pathway have been found to be associated with the disease states and altered cellular function (Han et al., 2020), and SubtypeDrug was subsequently developed to identify subtype-specific drugs at the subpathway (SP) level (Han et al., 2021). Although SubtypeDrug has identified some cancer candidate drugs, it did not consider the crosstalk between SPs. Actually, at the system biology level, the pathways/subpathways are not isolated, but crosstalk with each other.
Pathway crosstalk, which refers to the phenomenon of interaction among pathways or to the gene overlap among pathways, may provide the necessary insights into linked cellular processes and can further our understanding of alterations of biological states (Han et al., 2015; Sheng et al., 2021). Huang et al. (2019) presented that pathway crosstalk was a critical aspect of tumor pathways that allows cancer cells to acquire resistance to drug therapy, resulting in treatment failure. Li et al. (2008) developed a computational approach to detect crosstalk among pathways based on protein interactions between the pathway components. Donato et al. (2013) conducted a novel pathway-identification analysis by correcting crosstalk effects. More importantly, Csermely et al. (2013) proposed that numerous drug developmental failures were caused by undiscovered or underestimated pathway crosstalk effects. Thus, it is indispensable to develop a drug repurposing approach to identify cancer candidate drugs more efficiently by applying the crosstalk between pathways/subpathways.
Up to now, although there are many computational drug repurposing approaches available, all these approaches did not consider the crosstalk between pathways. Here, we developed a novel tool package called DRviaSPCN for drug repurposing by considering drug-induced SPs and their crosstalk effects. DRviaSPCN has three main functions (Fig. 1): (A) constructing an SP network based on the shared biological functions between SPs and calculating centrality scores of SPs in the context of gene expression data to reflect the influence of SP crosstalk; (B) evaluating drug–disease reverse association based on disease- and drug-induced SPs weighted by the SP crosstalk; (C) identifying cancer candidate drugs through perturbation analysis. This package also provides several visualization functions to illustrate the results.
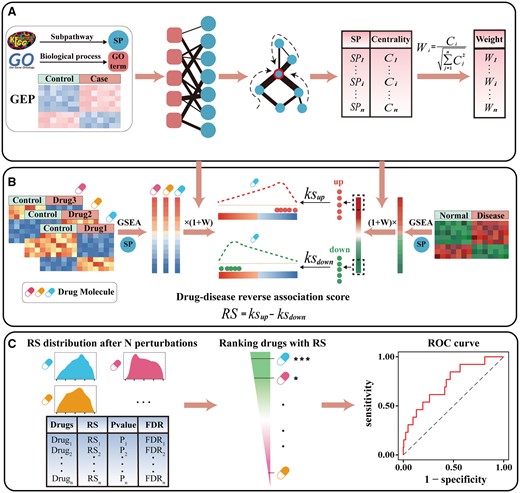
Flow diagram of DRviaSPCN.
2. Materials and methods
2.1 Constructing SP network and calculating centrality scores of SPs
We firstly downloaded all biological pathways from the KEGG database in XML format (Kanehisa et al., 2016) and constructed the pathway network with genes as nodes for each pathway based on pathway structure information (e.g. interactions, regulation, modifications and binding, between genes). Then, we used the SubpathwayMiner approach (Li et al., 2009, 2013) to extract the SPs from each KEGG pathway. This approach applies a k-clique algorithm to mine SPs according to pathway structure information, which will ensure that the genes in the SPs have highly similar biological functions. Considering that there may be some genes overlap between the SPs, we then used the algorithm proposed by Haider et al. (2018) to remove the redundant SPs. For each pair of SPs, if the genes overlap by more than 80%, the smaller SP will be discarded. Finally, 689 SPs were obtained after the elimination of redundancy.
The biological processes compiled in the Gene Ontology (GO) were then downloaded from MSigDB (Subramanian et al., 2005). The GO terms of biological processes with more than five genes and less than 100 genes remained, and 5548 biological processes were obtained. The drug-induced gene expression data of human cancer cell lines were downloaded from the CMap database (Lamb et al., 2006), and 3724 GEP of different cancer cell lines treated by the drugs with different concentrations and duration were established.
With these data, a drug-induced weighted network graph representation of SPs was constructed, which reflects the inherent interdependency among the SPs defined by cells, through their participation in common biological processes (GO terms) and augmented with measurements of transcriptional dysregulation specific to a pair of binary conditions (e.g. drug/control). In the network, SP nodes are more central, the more likely they are to be influenced by the crosstalk of transcriptionally altered SPs. We then calculated the centrality score for each SP to reflect their latent crosstalk influence in the network by using the random walk with restart algorithm (see Supplementary Materials). Finally, the centrality scores of SPs were normalized to a weight vector of SPs (Fig. 1A and Supplementary Figure S1).
2.2 Evaluating drug–disease reverse association
At the system biology level, the activities of SPs may be altered by drugs through not only the genes involved in the SPs but also the crosstalk between SPs. To identify the SPs regulated by a given drug, the genes in each SP were mapped to the list of differentially expressed genes regulated by the drug, and Gene Set Enrichment Analysis (Subramanian et al., 2005) was used to calculate the enrichment score (ES) of SP, which were then weighted with their latent crosstalk influence (normalized centrality score). According to the weighted ESs of SPs, a ranked SP list was formed for the drug (Fig. 1B).
To determine if the drug could potentially treat a given disease, a drug–disease reverse association score (RS) was defined to reflect the treatment extent of the drug at the SP level. For the disease, we performed a similar process to calculate the ESs of SPs and weighted them with the latent crosstalk influence calculated from the disease-induced SP network. The weighted ESs reflect the dysregulated extent of SPs induced by the disease. The up- and down-regulated SPs associated with the disease were respectively mapped to the ranked SP list regulated by a specific drug, and the Kolmogorov–Smirnov-like statistic was used to calculate RS (Supplementary Materials).
2.3 Identifying cancer candidate drugs
The greater negative RS indicates the drug may treat the disease to a larger extent, while the positive score indicates the drug may potentially promote disease development. Generally, a drug was applied with different instances (different concentrations, cancer cell lines or duration), we thus calculated the RS for each instance of all drugs. A descended ranked instance list was constructed according to the RSs of instances. For a given drug, the instance set of the drug was mapped to the ranked list, and the Kolmogorov–Smirnov statistic was used to compute the drug enrichment score (DES) based on the instance set. If the instance set of the drug enriches at the negative RS region of the list, the DES will be strong negative indicating the drug in different instances may have a consistent treatment effect on the disease. The statistical significance (P-value) of DES for the drug was estimated by comparing the observed DES of the drug with an empirical null distribution derived from randomly selecting the same number of instances for the drug (Fig. 1C), which was then adjusted using the false discovery rate approach proposed by Benjamini and Hochberg (Supplementary Materials).
3. Conclusion
The essential data used and produced by DRviaSPCN, which includes SP data, GO terms and SP enrichment scores induced by each drug, etc. have been deposited in our DRviaSPCNData package (https://github.com/hanjunwei-lab/DRviaSPCNData). We applied our approach to breast cancer data to show the predictive power, robustness analyses and comparison with other approaches. Supplementary Materials show the detailed data processing, method explanation and R code. In conclusion, DRviaSPCN is an efficient R-based tool that enables the identification of potential therapeutic drugs for cancer at the SP level via an SP crosstalk network. It may complement the recent computational drug repurposing approaches and contribute to the development of drug discovery.
Funding
This work was supported in part by the National Natural Science Foundation of China [62072145], the Natural Science Foundation of Heilongjiang Province [LH2019C042].
Conflict of Interest: none declared.
Data Availability
The data underlying this article are available in the article and in its online supplementary material.
References
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.


{kind=link}