





Abstract
Cyclization is a common strategy to enhance the therapeutic potential of peptides. Many cyclic peptide drugs have been approved for clinical use, in which the disulfide-driven cyclic peptide is one of the most prevalent categories. Molecular docking is a powerful computational method to predict the binding modes of molecules. For protein-cyclic peptide docking, a big challenge is considering the flexibility of peptides with conformers constrained by cyclization.
Integrating our efficient peptide 3D conformation sampling algorithm MODPEP2.0 and knowledge-based scoring function ITScorePP, we have proposed an extended version of our hierarchical peptide docking algorithm, named HPEPDOCK2.0, to predict the binding modes of the peptide cyclized through a disulfide against a protein. Our HPEPDOCK2.0 approach was extensively evaluated on diverse test sets and compared with the state-of-the-art cyclic peptide docking program AutoDock CrankPep (ADCP). On a benchmark dataset of 18 cyclic peptide-protein complexes, HPEPDOCK2.0 obtained a native contact fraction of above 0.5 for 61% of the cases when the top prediction was considered, compared with 39% for ADCP. On a larger test set of 25 cyclic peptide-protein complexes, HPEPDOCK2.0 yielded a success rate of 44% for the top prediction, compared with 20% for ADCP. In addition, HPEPDOCK2.0 was also validated on two other test sets of 10 and 11 complexes with apo and predicted receptor structures, respectively. HPEPDOCK2.0 is computationally efficient and the average running time for docking a cyclic peptide is about 34 min on a single CPU core, compared with 496 min for ADCP. HPEPDOCK2.0 will facilitate the study of the interaction between cyclic peptides and proteins and the development of therapeutic cyclic peptide drugs.
Supplementary data are available at Bioinformatics online.
1 Introduction
Peptides play an essential role in fundamental cellular functions by interacting with different proteins, such as signal transmission, gene expression, regulation and immunization (Araste et al., 2018; Lau and Dunn, 2018). The good target specificity, reduced toxicity and other favorable properties make peptides be attractive drug candidates, and thus the therapeutic potential of peptides has attracted high interest (Drucker, 2020; Lee et al., 2019; London et al., 2010). Over 60 peptide drugs have been approved to treat various diseases, including metabolic disease, oncology and cardiovascular disease (Lau and Dunn, 2018). Cyclization is a common modification to enhance the pharmacological properties of a peptide (Vu et al., 2021). Compared with linear peptides, cyclic peptides have several advantages like stability, easy production and better cell permeability (Craik and Du, 2017; Sanner et al., 2021). So far, more than 40 cyclic peptides have been approved for clinical use, in which the disulfide-driven cyclic peptide is one of the most prevalent categories (Jing and Jin, 2020; Zorzi et al., 2017). The oncology drugs lanreotide and romidepsin are examples of such disulfide-driven cyclic peptides (Zorzi et al., 2017).
The structure of protein–peptide complexes is valuable for understanding their interactions and rationally designing new drugs. Experiments like X-ray and Nuclear Magnetic Resonance (NMR) suffer from high costs and technical difficulties, which promotes the development of various computational methods (Duffy et al., 2019; Nguyen et al., 2018; Ru and Lin, 2021; Taherzadeh et al., 2018; Yu and Lin, 2015; Zhao et al., 2018). Molecular docking is a powerful approach to predict the binding modes between two molecules. For protein–peptide docking, the inherent flexibility of peptides is a big challenge due to the more rotatable bonds in peptides than small molecules, where the conformational space of a molecule grows exponentially with its number of rotatable bonds. For linear peptide docking, several docking and refinement protocols have been developed to predict protein–peptide interactions (Ben-Shimon and Niv, 2015; Lee et al., 2015; Pallara et al., 2017; Trellet et al., 2013; Weng et al., 2020; Yan et al., 2016). The pepATTRACT protocol (de Vries et al., 2017; Schindler et al., 2015) uses a coarse-grained rigid-body docking search followed by a flexible atomic refinement. The CABS-dock protocol (Kurcinski et al., 2015) utilizes Replica Exchange Monte Carlo dynamics and the k-medoids clustering. PIPER-FlexPepDock (Alam et al., 2017) integrates the Rosetta fragment picker and PIPER docking to generate peptide fragment ensembles followed by rigid docking against receptors. The AutoDock CrankPep (ADCP) protocol (Zhang and Sanner, 2019a) adopts CRANKITE (Podtelezhnikov and Wild, 2008) to perform Monte Carlo (MC) searches of peptide binding modes within AutoDock affinity grids (Huey et al., 2007). Compared with the advancement in linear peptide docking, docking approaches for cyclic peptides still fall behind. The cyclic conformation of peptides presents unique challenges for computational prediction. Monte Carlo (Zhang and Sanner, 2019b) and molecular dynamics simulations (Damjanovic et al., 2021; Kato et al., 2017), including replica-exchange molecular dynamics (Qi et al., 2018; Zhou, 2007), Metadynamics (Laio and Parrinello, 2002; Yu and Lin, 2015) and Bias-Exchange Metadynamics (Piana and Laio, 2007) may be used to generate the conformational ensembles of cyclic peptides. The new version of ADCP (Zhang and Sanner, 2019b) introduces a backbone cyclization potential and a distance-dependent potential for disulfide bonds in Monte Carlo searches to dock peptides cyclized through backbones or disulfide bonds. However, these methods are time-consuming and require many computing resources. Ensemble docking is a powerful strategy to consider molecular flexibility with high-computational efficiency, which first generates an ensemble of peptide conformations and then performs rigid docking against the protein receptor.
Recently, we have developed a fast hierarchical flexible peptide docking algorithm HPEPDOCK (Tao et al., 2020; Zhou et al., 2018a, b) to predict the binding modes between linear peptides and proteins. HPEPDOCK achieved an excellent performance through the efficient generation and ensemble docking of peptide conformations. Given the therapeutic potential of cyclic peptides, here we introduce a new version of HPEPDOCK, named HPEPDOCK2.0, to support the efficient prediction of the binding modes between proteins and peptides cyclized through a disulfide bond. By taking advantage of our efficient peptide three-dimensional conformation sampling algorithm MODPEP2.0 (Yan et al., 2017) and the knowledge-based scoring function ITScore-PP (Huang and Zou, 2008), HPEPDOCK2.0 achieved a good performance on diverse test sets in both bound and unbound docking.
2 Materials and methods
HPEPDOCK2.0 is an improved version of HPEPDOCK, which can additionally handle peptides cyclized through a disulfide bond. Same as the original version, HPEPDOCK2.0 considers the peptide flexibility through ensemble docking of multiple peptide conformations. Given a protein structure and a peptide sequence with the disulfide bond information, HPEPDOCK2.0 predicts the binding modes of the protein–peptide complex through two stages: sampling of peptide conformations and docking of the modeled peptide conformations against the protein, which are explained in details as follows.
2.1 Peptide conformation sampling
Our MODPEP program (Yan et al., 2017) can fast generate the conformations of a linear peptide from a sequence. Inspired by the therapeutic potential of cyclic peptides, we have developed an extended version, named MODPEP2.0, to generate the peptide 3D conformations of cyclic peptides formed by a disulfide bond (Tao et al., 2022). Specifically, given a sequence of the peptide and the disulfide bond information, MODPEP2.0 reads the cyclic backbone library and selects a backbone template of the cyclic fragment according to the probability, which depends on the sequence similarity between the target sequence and the template sequence and the resolution of the original template. Except for Cysteine, which contains the backbone and side chain, other residues in the cyclic template library only have the backbone. This is because the side chain of Cysteine is the key for disulfide bonds, which directly leads to the formation of a cyclic peptide. With the selected cyclic backbone template, MODPEP2.0 randomly selects a rotamer from the single-residue rotamer library to construct the corresponding side chain of other amino acids. The residues outside the cyclic fragment were built the same way as the previous version to model linear peptides (Yan et al., 2017). Specifically, according to the secondary structure information predicted by PSIPRED (Jones, 1999), MODPEP2.0 adds the helix or sheet fragments of the corresponding length or expands coil residues one by one on the current structure. The newly added fragment or residue will be checked to see whether there is an atomic clash with the existing peptide structure. If there exist severe clashes, the fragment or residue will be reselected and rebuilt. Notably, if there is no suitable fragment or residue structure in the libraries, MODPEP2.0 will return to the previous step to remodel the previous residue. The above process is repeated until all residues of the target sequence are built. A variety of structures in the cyclic backbone library and other linear backbone libraries ensure the diversity of final peptide conformers. MODPEP2.0 has been evaluated in a non-redundant dataset consisting of 193 diverse cyclic peptides with peptide lengths ranging from 5 to 30, and showed high accuracy with the average Cα Root Mean Square Deviation (RMSD) of 2.20 and 1.66 Å when the top 10 and 100 conformations were considered. Like the original version, MODPEP2.0 is also efficient and can model 100 peptide structures in 1 s. The number of peptide conformations generated for ensemble docking is 1000 by default.
2.2 Docking algorithm
The docking protocol for HPEPDOCK2.0 is the same as that for HPEPDOCK. Namely, it uses a modified version of MDock (Huang and Zou, 2007) to dock the peptide conformations against the receptor protein structure. Here, the protein and peptides are treated as rigid bodies, and the peptide flexibility is considered by an ensemble of different conformations generated by MODPEP2.0 mentioned above. First, the DMS program (Richards, 1977) is used to generate the molecular surface of the protein. Next, the normal vector at each surface point is computed. Then, the SPHGEN program (Ewing et al., 2001) is used to generate sets of sphere points representing the negative image of binding pockets on the protein according to the molecular surface and the normal vectors. The putative binding modes of peptides are sampled by matching the peptide atoms with the sphere points (Ewing and Kuntz, 1997). It is worth noting that we have modified MDock to use a reduced model of the peptide during docking. Namely, only the Cα atom and the center of mass of the side chain for each residue are kept to match the sphere points, so as to reduce the memory consumption and speed up the sampling process. The ligand orientation that matches more sphere points was first selected for further binding energy score evaluation. During scoring, the reduced models of peptides are replaced by their all-atom models. ITScorePP (Huang and Zou, 2008) is used to calculate the protein–peptide binding score of sampled binding modes instead of the original protein-ligand scoring function of MDock. ITScorePP is an efficient knowledge-based energy scoring function for protein–protein interactions. A SIMPLEX minimization algorithm (Nelder and Mead, 1965) is used to minimize the binding energy of each peptide orientation. Finally, different peptide orientations are ranked according to their binding energy scores and clustered using 1.0 Å rmsd cutoff. The one with lower energy in a cluster is kept.
2.3 Test sets
A disulfide-driven cyclic peptide dataset generated by Zhang and Sanner (2019b) was used to test our cyclic peptide docking approach on bound receptor structures. Two complexes in this dataset containing multiple disulfide bonds in the peptide were excluded, resulting in a final set of 18 complexes, named TS18. It consists of 18 non-redundant protein–peptide complexes with peptide lengths ranging from 6 to 20 residues. The complexes in the benchmark are all X-ray crystal structures with a resolution of 2.2 Å or better. The complexes in TS18 are the representative ones in each cluster at 70% sequence identity of the receptor. The receptor structures used for docking in TS18 are the experimental native structures. In addition, out of the 18 complexes in TS18, there are 10 test cases whose protein has an experimental apo structure (Zhang and Sanner, 2019b). These 10 cases with experimental apo receptors, named TS10_apo, were used for unbound docking evaluations. It is known that docking peptides to unbound receptors is more realistic but also more challenging. These two datasets have been used to evaluate ADCP for cyclic peptide docking (Zhang and Sanner, 2019b). Therefore, the docking results for ADCP are available on these datasets, which can be used as references to assess our docking algorithm.
For further evaluations, we have also built a new test set containing experimental native receptor structures. First, we searched the Protein Data Bank (PDB) (Berman, 2000; Burley et al., 2017) to select the protein-cyclic peptide complexes with resolution better than 4.0 Å and the peptides containing 5–30 standard amino acids. Then, we used CD-HIT (Li and Godzik, 2006) to cluster the sequences of these peptides, and the one with the best resolution was retained. Peptides containing long linear parts were excluded. Namely, peptides with a cyclic length less than one-third of the entire length were removed. Then, we clustered the receptor sequence by CD-HIT with a 70% sequence identity threshold and selected a representative complex per cluster. Finally, the overlapped complexes with those of TS18 were removed to prevent redundancy, leading to a final set of 25 protein–peptide complexes, called TS25. The detailed list of the 25 complexes is shown in Supplementary Table S1.
In addition, the crystal structure of the receptor may be not available in real applications. In such cases, the 3D structure of the receptor needs to be predicted. Therefore, to test the performance of HPEPDOCK2.0 on predicted receptor structures, we have used the well-known protein structure prediction method AlphaFold2 (AF2; Jumper et al., 2021) to model the structures of the receptors in TS18. As AF2 is a deep learning approach developed for structure prediction of protein monomers, we excluded those cases with multiple protein chains in the receptor, yielding a final subset of 11 protein–peptide complexes. To mimic a realistic situation, all the protein structures that were published on or after the released date of the test case were excluded from the templates. All the default parameters were used during the modeling with AF2. The sequences of the receptor were extracted from the SEQRES lines of the PDB file (Tsaban et al., 2022). The top-ranked model predicted by AF2 for each receptor was used. AF2 provides residue-level pLDDT values as a confidence estimate for each model. We removed the residues with low accuracy using a pLDDT cutoff of 60 and found that those residues are usually located in loops with high flexibility or long coils in the head or tail. Here, we named this test set with predicted receptor structures TS11_AF2 to distinguish it from other datasets mentioned above.
2.4 Evaluation criteria
For easy reference and comparison, we adopted the same success metric as that for ADCP to evaluate the docking performance of HPEPDOCK2.0. Namely, the fraction of native contacts () reproduced in a predicted docking pose was used for measuring the quality of a predicted protein–peptide binding mode (Méndez et al., 2003). Here, the native contacts were calculated based on the residue pairs between the protein and the peptide that are within 5 Å from each other in the native structure. Similar to the criteria in the Critical Assessment of Prediction of Interactions (CAPRI) experiments (Janin, 2010; Janin et al., 2003), an of above 0.3 is considered as the medium quality and an of above 0.5 is considered the high quality. In this study, if a binding mode has an of above 0.5, it was considered a successful prediction. The success rate was defined as the percentage of the cases with at least one successful prediction when a certain number of top predictions were considered in each test set.
3 Results and discussion
3.1 Performance of HPEPDOCK2.0
To evaluate the ability of HPEPDOCK2.0 in predicting the binding modes of protein-cyclic peptide complexes, we have docked the cyclic peptides into their bound, apo or predicted receptor structures in the four test sets mentioned above. The docking results of HPEPDOCK2.0 on the four datasets are presented and discussed in the following. It is noteworthy that we have removed the corresponding sequence of the cyclic peptide in the cyclic backbone library to eliminate the redundancy when sampling the conformations of a cyclic peptide by MODPEP2.0.
3.1.1 Performance on TS18 with bound receptor structures
Table 1 and Figure 1 show the docking performance of HPEPDOCK2.0 on the test set of 18 complexes in TS18 when several numbers of top predictions were considered. For comparison, Table 1 and Figure 1 also give the docking results of ADCP, which were taken from Zhang and Sanner (2019b). The detailed native contact fractions of the predicted binding modes are listed in Supplementary Table S2. It can be seen from Table 1 and Figure 1 that HPEPDOCK2.0 achieved a better performance than ADCP, especially when only the top-ranked pose was considered. According to evaluation criteria, HPEPDOCK2.0 obtained a success rate of 61% when the top docking mode was considered, which is a significant improvement compared to 39% for ADCP. HPEPDOCK2.0 yielded a better performance by reproducing an average native contact fraction of 0.57 for the top 1 binding mode, 0.64 for the top 5 predictions and 0.67 for the top 10 predictions, compared with 0.44, 0.57 and 0.62 for ADCP. Notably, HPEPDOCK2.0 showed a better scoring capability by ranking the solutions with within the top 1 for 11 targets (61%). It can also be seen from Supplementary Table S2 that the of docked pose tends to decrease with the increase of peptide length, which may be attributed to the larger sampling space of binding conformations and orientations of long peptides.
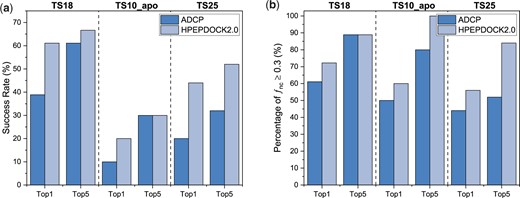
Comparison of the docking performances between HPEPDOCK2.0 and ADCP for top 1 and 5 predictions on the three test sets of TS18, TS10_apo and TS25. (A) Success rate under the criterion of . (B) Percentage of the test cases under the criterion of
Performances of HPEPDOCK2.0 and ADCP in docking cyclic peptides on the test sets of TS18 (bound receptor structures), TS10_apo (apo receptor structures), TS25 (bound receptor structures) and TS11_AF2 (predicted receptor structures) when the top 1, 5, 10 and 100 predictions were considered
| Test set | Metric | ADCP | HPEPDOCK2.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Top 1 | Top 5 | Top 10 | Top 100 | Top 1 | Top 5 | Top 10 | Top 100 | ||
| TS18 | Avg. | 0.44 | 0.57 | 0.62 | 0.71 | 0.57 | 0.64 | 0.67 | 0.73 |
| 11 | 16 | 17 | 18 | 13 | 16 | 17 | 18 | ||
| 7 | 11 | 14 | 16 | 11 | 12 | 14 | 16 | ||
| TS10_apo | Avg. | 0.29 | 0.46 | 0.53 | 0.64 | 0.42 | 0.52 | 0.57 | 0.68 |
| 5 | 8 | 10 | 10 | 6 | 10 | 10 | 10 | ||
| 1 | 3 | 5 | 6 | 2 | 3 | 5 | 8 | ||
| TS25 | Avg. | 0.28 | 0.35 | 0.40 | 0.48 | 0.45 | 0.55 | 0.61 | 0.70 |
| 11 | 13 | 16 | 17 | 14 | 21 | 22 | 25 | ||
| 5 | 8 | 10 | 12 | 11 | 13 | 17 | 23 | ||
| TS11_AF2 | Avg. | – | – | – | – | 0.37 | 0.45 | 0.53 | 0.63 |
| – | – | – | – | 6 | 8 | 9 | 10 | ||
| – | – | – | – | 3 | 4 | 5 | 7 | ||
| Test set | Metric | ADCP | HPEPDOCK2.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Top 1 | Top 5 | Top 10 | Top 100 | Top 1 | Top 5 | Top 10 | Top 100 | ||
| TS18 | Avg. | 0.44 | 0.57 | 0.62 | 0.71 | 0.57 | 0.64 | 0.67 | 0.73 |
| 11 | 16 | 17 | 18 | 13 | 16 | 17 | 18 | ||
| 7 | 11 | 14 | 16 | 11 | 12 | 14 | 16 | ||
| TS10_apo | Avg. | 0.29 | 0.46 | 0.53 | 0.64 | 0.42 | 0.52 | 0.57 | 0.68 |
| 5 | 8 | 10 | 10 | 6 | 10 | 10 | 10 | ||
| 1 | 3 | 5 | 6 | 2 | 3 | 5 | 8 | ||
| TS25 | Avg. | 0.28 | 0.35 | 0.40 | 0.48 | 0.45 | 0.55 | 0.61 | 0.70 |
| 11 | 13 | 16 | 17 | 14 | 21 | 22 | 25 | ||
| 5 | 8 | 10 | 12 | 11 | 13 | 17 | 23 | ||
| TS11_AF2 | Avg. | – | – | – | – | 0.37 | 0.45 | 0.53 | 0.63 |
| – | – | – | – | 6 | 8 | 9 | 10 | ||
| – | – | – | – | 3 | 4 | 5 | 7 | ||
Performances of HPEPDOCK2.0 and ADCP in docking cyclic peptides on the test sets of TS18 (bound receptor structures), TS10_apo (apo receptor structures), TS25 (bound receptor structures) and TS11_AF2 (predicted receptor structures) when the top 1, 5, 10 and 100 predictions were considered
| Test set | Metric | ADCP | HPEPDOCK2.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Top 1 | Top 5 | Top 10 | Top 100 | Top 1 | Top 5 | Top 10 | Top 100 | ||
| TS18 | Avg. | 0.44 | 0.57 | 0.62 | 0.71 | 0.57 | 0.64 | 0.67 | 0.73 |
| 11 | 16 | 17 | 18 | 13 | 16 | 17 | 18 | ||
| 7 | 11 | 14 | 16 | 11 | 12 | 14 | 16 | ||
| TS10_apo | Avg. | 0.29 | 0.46 | 0.53 | 0.64 | 0.42 | 0.52 | 0.57 | 0.68 |
| 5 | 8 | 10 | 10 | 6 | 10 | 10 | 10 | ||
| 1 | 3 | 5 | 6 | 2 | 3 | 5 | 8 | ||
| TS25 | Avg. | 0.28 | 0.35 | 0.40 | 0.48 | 0.45 | 0.55 | 0.61 | 0.70 |
| 11 | 13 | 16 | 17 | 14 | 21 | 22 | 25 | ||
| 5 | 8 | 10 | 12 | 11 | 13 | 17 | 23 | ||
| TS11_AF2 | Avg. | – | – | – | – | 0.37 | 0.45 | 0.53 | 0.63 |
| – | – | – | – | 6 | 8 | 9 | 10 | ||
| – | – | – | – | 3 | 4 | 5 | 7 | ||
| Test set | Metric | ADCP | HPEPDOCK2.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Top 1 | Top 5 | Top 10 | Top 100 | Top 1 | Top 5 | Top 10 | Top 100 | ||
| TS18 | Avg. | 0.44 | 0.57 | 0.62 | 0.71 | 0.57 | 0.64 | 0.67 | 0.73 |
| 11 | 16 | 17 | 18 | 13 | 16 | 17 | 18 | ||
| 7 | 11 | 14 | 16 | 11 | 12 | 14 | 16 | ||
| TS10_apo | Avg. | 0.29 | 0.46 | 0.53 | 0.64 | 0.42 | 0.52 | 0.57 | 0.68 |
| 5 | 8 | 10 | 10 | 6 | 10 | 10 | 10 | ||
| 1 | 3 | 5 | 6 | 2 | 3 | 5 | 8 | ||
| TS25 | Avg. | 0.28 | 0.35 | 0.40 | 0.48 | 0.45 | 0.55 | 0.61 | 0.70 |
| 11 | 13 | 16 | 17 | 14 | 21 | 22 | 25 | ||
| 5 | 8 | 10 | 12 | 11 | 13 | 17 | 23 | ||
| TS11_AF2 | Avg. | – | – | – | – | 0.37 | 0.45 | 0.53 | 0.63 |
| – | – | – | – | 6 | 8 | 9 | 10 | ||
| – | – | – | – | 3 | 4 | 5 | 7 | ||
Figure 2 shows four examples of protein–peptide complexes in the top 10 predictions, two of which are successfully docked and two failed. For 5djc and 5wxr, the predicted peptide binding conformation overlaps with the native crystal structure well, and almost all native contacts have been reproduced. However, HPEPDOCK2.0 failed to predict the binding modes for 3p72 and 4m1d within the top 10 predictions. The failure may be attributed to several reasons. First, one key contributor to the failure is the poor quality of sampled peptide conformations. The backbone RMSDs between the peptide conformations and native structures are 3.19 and 4.53 Å for 3p72 and 4m1d (Fig. 2C and D), respectively. For 3p72, the secondary structure is critical, but there are few similar backbones in the cyclic backbone library. As such, MODPEP2.0 is not able to model this secondary structure due to the lack of templates. Second, the deep and narrow groove in the binding site increases the difficulty of docking. The native structure fits well in the groove, as shown in Figure 2D for 4m1d, but it is difficult for the sampled conformations to fit into such narrow groove well. It can be seen from Supplementary Table S2 that the better docking results are ranked low for 4m1d. In addition, the scoring function ITScore_PP developed from protein–protein interactions still has room for improvement of protein–peptide docking. These reasons provide insights into further improvement of our docking program. Developing a scoring function for protein–peptide interactions and a better cyclic peptide conformation sampling program may further improve the docking performance.
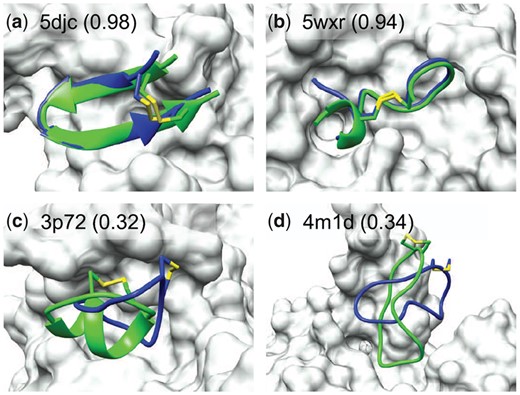
Comparison between the crystal structure (green) and predicted peptide binding modes by HPEPDOCK2.0 (blue) for two successful and two failed examples when the top 10 predictions were considered on the TS18 test set. The protein is represented by molecular surface (grey) and the fraction of native contacts reproduced by the docked pose is listed in the parentheses (A color version of this figure appears in the online version of this article.)
3.1.2 Performance on TS10_apo with apo receptor structures
Table 1 and Figure 1 report the docking performance of HPEPDOCK2.0 on the 10 apo receptor structures in TS10_apo when several numbers of top predictions were considered. For comparison, the data of ADCP from Zhang and Sanner (2019b) are also given in the table and figure. The detailed native contact fractions of the predicted binding modes are listed in Supplementary Table S3. It can be seen from Table 1 and Figure 1 that HPEPDOCK2.0 obtained a less satisfactory performance on TS10_apo than on TS18, which reflects the impact of protein conformational changes on docking. It is known that the receptors tend to have induced conformational changes when binding with a peptide. For TS10_apo test set, we directly docked different cyclic peptide conformations generated by MODPEP2.0 to the apo protein structures without considering the flexibility of proteins. Predicting the induced conformational changes of receptors is challenging, which is a future direction to improve our HPEPDOCK. Nevertheless, HPEPDOCK2.0 also achieved a significantly better docking performance than ADCP (Table 1 and Figure 1). On average, HPEPDOCK2.0 obtained a native contact fraction of when the first prediction was considered, which is 0.13 higher than 0.29 for ADCP. The cases with for HPEPDOCK2.0 were more or the same as those for ADCP. If considering the top five solutions, HPEPDOCK2.0 predicted 3 complexes out of 10 with , and the other 7 targets with , compared with 3 and 5 cases for ADCP, respectively. Compared with the ranks of the first successful predictions on TS18, the corresponding ranks on TS10_apo were worse (Supplementary Table S3). This can be understood because our scoring function calculates the binding energy of all-atom intermolecular interactions. The uncertainties of side chains within the binding pocket of the apo structure would result in inaccurate binding scores of predicted binding modes. This is a general challenge for the scoring function to rank the docking poses of rigid docking within apo receptor structures.
3.1.3 Performance on TS25 with bound receptor structures
Table 1 and Figure 1 show the docking accuracies of HPEPDOCK2.0 on the test set of 25 complexes in TS25 when several numbers of top predictions were considered. For comparison, we also calculated the docking results of ADCP on the TS25 set. Specifically, we downloaded the ADCP software and docked the peptide against its receptor using default docking parameters and 50 independent replicas with 2.5 million scoring evaluations each. The docking box was defined using the native peptide structure with a padding of 4 Å added to every side of the bounding box (Zhang and Sanner, 2019b). The native protein structure and the peptide sequence were used for docking. ADCP introduced a distance-dependent potential between the sulfur atoms of cysteine residues to create or break disulfide bonds during simulations, instead of specifying the Cysteines to form disulfide bonds. The detailed native contact fractions of the predicted binding modes by HPEPDOCK2.0 and ADCP are listed in Supplementary Table S4. It can be seen from Table 1 and Figure 1 that HPEPDOCK2.0 obtained a relatively lower success rate on TS25 than on TS18, which would be due to the lower structure quality and the longer peptide sequence in TS25 than in TS18. Specifically, the complexes of TS25 have an overall lower resolution of <4.0 Å and longer peptides with 5–30 residues, compared with the resolution of Å and the peptides of 6–20 residues for TS18. Nevertheless, HPEPDOCK2.0 still yielded a much better performance than ADCP on the TS25 test set and predicted the binding modes with for 14 complexes when the top prediction was considered, compared with 11 cases for ADCP. Under the criterion of , HPEPDOCK2.0 achieved a success rate of 44% (11 out of 25 cases) when the top solution was considered, compared with 20% (5 out of 25 cases) for ADCP. Similar trends can also be observed for the top 5, 10 and 100 predictions (Table 1). Another feature in Supplementary Table S4 is that the peptides with more residues tend to have lower accuracy. The failure of ADCP would be due to the limited number of Monte Carlo replicas and scoring evaluation steps. According to the ADCP tutorial, longer peptides require more independent Monte Carlo searches and steps, although we used the default parameters for different complexes to provide a fair reference.
Figure 3 shows the predicted docking poses of six examples by HPEPDOCK2.0 when the top 10 predictions were considered. It can be seen from the figure that HPEPDOCK2.0 accurately predicts the binding modes of some complexes and yielded a high native contact fraction of above 0.9 for the docked poses (Fig. 3A–C). However, HPEPDOCK2.0 failed to give a binding mode with on some complexes such as 6uib, 5jz8 and 6cbp (Fig. 3D–F). It can be seen from Figure 3D that the predicted position and direction of the peptide are roughly similar to the native ones, but the major difference in the secondary structure may cause the failure. As for 5jz8 in Figure 3E, the binding pocket includes a deep and long channel and a narrow internal part, which leads to extremely high requirements for the conformation of the peptide. A slight difference in the peptide structure may cause severe atomic clashes and push the peptide away from the real binding position. The failure of 6cbp may be due to its long peptide of 30 residues, which is the longest in all test cases. It can be seen from Figure 3F that the binding position of the peptide is different from that of the crystal structure, and the binding direction is opposite to the native one. In addition, this case also has an open binding site, which will allow peptides to adopt multiple binding positions. It is challenging for a scoring function to select the correct binding modes which have good contacts with the protein.
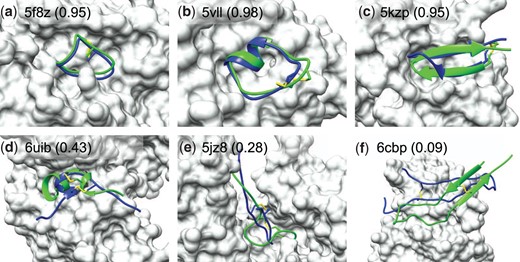
Comparison between the crystal structure (green) and predicted peptide binding modes by HPEPDOCK2.0 (blue) for three successful and three failed examples when the top 10 predictions were considered on TS25. The protein is represented by molecular surface (grey) and the fraction of native contacts reproduced by the docked pose is listed in the parentheses (A color version of this figure appears in the online version of this article.)
The disulfide bond information used in the test is from the native peptide. If there are only two Cysteines in the sequence, the residues to form the disulfide bond may be determined directly. However, the information about the disulfide bond may be uncertain when more than two Cysteines are in a peptide. There are two peptides in TS25, chain B of 5JZ8 and chain I of 3SSB, which contain more than two Cysteines. In such cases, users may need to predict the disulfide bond with a third-party program. As an example, here we used Dlpro2.0 to predict the disulfide connectivity pattern of 5JZ8_B and 3SSB_I. Dlpro2.0 is an integrated, modular approach for disulfide bridge predictions using kernel methods, 2D recursive neural networks and weighted graph matching (Cheng et al., 2006). The true disulfide bond information of 5JZ8_B was successfully predicted by Dlpro2.0. However, there are five Cysteines in 3SSB_I, two of which form an intra-disulfide bond in the peptide and three form inter-disulfide bonds with the receptor. It is difficult to predict the real disulfide bond without considering the protein of the peptide to bind. It needs to be emphasized that the covalent bond between the receptor and the ligand is not considered in HPEPDOCK2.0, and the performance on 3SSB_I is to provide a performance reference of peptides with many Cysteines. Dlpro2.0 wrongly predicted two disulfide bonds of 3–21 and 7–27, but the real pair is 9–21. The incorrect disulfide bond information led to a deviation between the peptide conformations generated by MODPEP20 and the native peptide structure. Nevertheless, HPEPDOCK2.0 still reproduced some native contacts with a fraction of up to 0.24 for the top 10 predictions, even though the wrong disulfide bond information was used.
3.1.4 Performance on TS11_AF2 with predicted receptor structures
Table 1 lists the docking results of HPEPDOCK2.0 on a test set of 11 protein–peptide complexes, named TS11_AF2, where the receptor structures are predicted by AlphFold2. The and rank of the first solution with by HPEPDOCK2.0 and the RMSDs of predicted receptor models are listed in Supplementary Table S5. It can be seen from the table that HPEPDOCK2.0 obtained a success rate of 45% when the top 10 binding modes were considered. On average, HPEPDOCK2.0 reproduced a native contact fraction of 0.37 for the top 1 prediction, 0.45 for the top 5 predictions and 0.53 for the top 10 predictions. Overall, HPEPDOCK2.0 yielded a better performance on the test sets of TS18 and TS25 with bound receptor structures than on the test set of TS10_apo with apo receptor structures and TS_AF2 with predicted receptor structures. The different performances of HPEPDOCK2.0 on the three categories of bound, apo and predicted receptor structures suggest the importance of the quality of the receptor structure in predicting accurate peptide binding modes.
Figure 4 shows four examples of docked protein–peptide complexes, including two successful ones and two failed ones, when the top 10 binding modes were considered. For 1smf, the protein structure predicted by AF2 and the peptide binding mode predicted by HPEPDOCK2.0 overlap with the native crystal structures well (Fig. 4A). For 5djc, although the backbone RMSD (bRMSD) between the predicted protein model and the experimental structure is as high as 3.45 Å, the docking pose predicted by HPEPDOCK2.0 still reproduced most of the native contacts (Fig. 4B). This may be due to the accurate prediction of the peptide binding pocket on the protein of 5djc. However, HPEPDOCK2.0 failed to predict an accurate binding mode on 5xco and 5vb9 (Fig. 4C and D). It can be seen from the figure that the predicted receptor models of 5xco and 5vb9 have some clashes with the native structure of peptides, blocking the binding pockets. HPEPDOCK2.0 performs rigid protein–peptide docking, without considering the flexibility of receptors, so it is difficult to dock the peptide into their suitable positions.
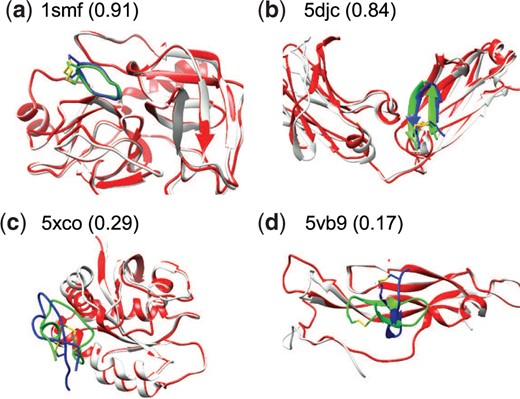
Comparison between the crystal structure (green) and predicted peptide binding modes by HPEPDOCK2.0 (blue) for two successful and two failed examples when the top 10 predictions were considered on the test set of TS11_AF2. The predicted protein model by AF2 (red) is superimposed onto the experimental bound structure (grey). The fraction of native contacts reproduced by the docked pose is listed in the parentheses (A color version of this figure appears in the online version of this article.)
3.2 Impact of the number of peptide conformations
By default, the number of peptide conformations used for ensemble docking is 1000 for a peptide. However, users may not use so many peptide conformations for docking calculation in some cases. Therefore, we explored the impact of the number of peptide conformations on the performance of HPEPDOCK2.0. Figure 5 shows the success rates as a function of the number of peptide conformations on the test sets of TS18 and TS25 when the top 1 and 10 predictions were considered. It can be seen from the figure that the success rates increase quickly in the first 50 peptide conformations and then gradually become stable with the increasing number of conformations. Generally, more conformations tend to yield a higher success rate, although there exist some fluctuations in the statistics of the success rate due to the small size of the test set. Overall, the success rate becomes stable after 500–600 conformations for the top prediction and reaches the maximum after 200 conformations for the top 10 predictions. Therefore, users may choose 50, 200 or 500 conformations per peptide in real application depending on their needs.
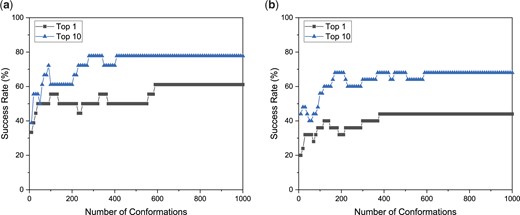
Average success rate of HPEPDOCK2.0 for the protein–peptide complexes on the test sets of TS18 (A) and TS25 (B) as a function of the number of peptide conformations when the top 1 and top 10 docking poses were considered
3.3 Computational efficiency
In addition to the good docking performance, HPEPDOCK2.0 also has a high-computational efficiency. The sampling program MODPEP2.0 can sample 1000 conformations in 1 min. The running time of HPEPDOCK2.0 is mainly spent on the docking step. For comparison, we also calculated the running time of ADCP. As ADCP uses multi-core to accelerate the docking calculation, we converted the ADCP multi-core time to single-core time. Figure 6 shows the average running time of docking cyclic peptides of different lengths on the TS25 dataset. The dotted line is the overall average time. HPEPDOCK2.0 docked 1000 conformations against the protein for each target, and ADCP performed 50 independent searches with 2.5 million evaluations each. The docking calculations were conducted on the CPU cores (Intel Xeon CPU E5-2690 v4 @ 2.60 GHz) on a Linux x86_64 cluster. It can be seen from Figure 6 that the running time of HPEPDOCK2.0 is much shorter than ADCP for all test cases. The average time of ADCP is 495.59 min on a single core, while HPEPDOCK2.0 only needs 34.29 min, which is about 1/14 of the former.
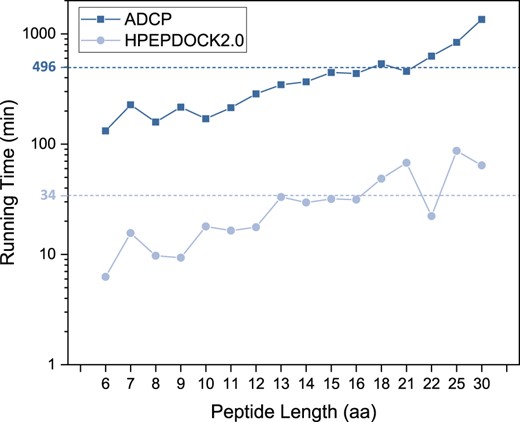
Average running time of HPEPDOCK2.0 and ADCP for docking cyclic peptides of different lengths on the TS25 set of 25 complexes. The overall average values of the running times for the 25 complexes are shown in dashed lines
The higher computational efficiency of HEPDOCK2.0 than ADCP can be attributed to the faster speed on both stages of the docking pipeline: sampling and scoring. For sampling, ADCP uses a Monte Carlo-based sampling of peptide conformations within the binding pocket, starting with an extended conformation of the peptide. A distance-dependent potential between the sulfur atoms of Cysteine residues is used to form the disulfide bond. Such conformational sampling of the peptide is computationally expensive. In contrast, HPEPDOCK2.0 directly docks an ensemble of cyclic peptide conformations by regular rigid peptide docking, which is much faster than Monte Carlo simulations. For scoring, each Monte Carlo step of ADCP requires an evaluation by the scoring function. That is to say, millions of scoring operations are in progress in each replica of ADCP. In contrast, HPEPDOCK2.0 scores thousands of putative binding modes and does optimizations with our fast knowledge-based scoring function ITScorePP.
From Figure 6, we can also see that both HPEPDOCK2.0 and ADCP tend to have a longer running time with the increase of peptide length and show an approximately exponential relationship with the peptide length. For ADCP, the exponentially increasing time is due to the exponentially increasing number of conformations with the peptide length in the MC simulations. For HPEPDOCK2.0, a key contributor to the exponential relationship is the exponential increase of the possible matches between the peptide and the sphere points during the generation of putative binding modes. Heuristic pruning algorithm to remove redundant matches and filter out unsuitable binding poses can be developed to accelerate peptide docking and prevent the exponential growth of running time in our future study. The detailed times are shown in Supplementary Table S6. HPEPDOCK2.0 can dock peptides within 12 residues in <20 min. For long peptides, HPEPDOCK2.0 tends to spend more time on orientational sampling and shape matching for narrow pockets. For example, the running time for 5jz8 (Fig. 3E) is about 54 min, which is higher than 27 min for 6cbp (Fig. 3F). This is because the much narrower pocket of 5jz8 needs more time to sample good candidates that pass atomic clash checking. In addition, the short running time for the peptide of length 22 in Figure 6 is due to the open binding pocket of 6gfq, which enables fast sampling of peptide binding modes.
4 Conclusions
We proposed a new version of HPEPDOCK, named HPEPDOCK2.0, which can support the docking of cyclic peptides through a disulfide bond. Same as the previous version, HPEPDOCK2.0 first samples an ensemble of peptide conformations by MODPEP2.0 and then docks the sampled conformations into the binding pocket of the protein. With the efficient sampling ability of MODPEP2.0 and high accuracy of docking algorithm MDock coupled with the knowledge-based scoring function ITScorePP, HPEPDOCK2.0 achieved an excellent performance on three diverse test sets, compared with the state-of-the-art docking program ADCP. Specifically, HPEPDOCK2.0 obtained the binding modes with for 11 targets (61% of all cases), compared with only 7 targets (39% of all cases) for ADCP when the top prediction was considered on the TS18 dataset of 18 complexes. Tested on TS10_apo and TS25, HPEPDOCK2.0 also yielded a better performance than ADCP. In addition, HPEPDOCK2.0 was also validated on a dataset with predicted receptor structures. HPEPDOCK2.0 is computationally efficient and the average running time on a single core is only 34 min, compared with 496 min for ADCP. It is expected that HPEPDOCK2.0 will facilitate the study of the interaction between cyclic peptides and proteins and the development of therapeutic cyclic peptide drugs.
Funding
This work is supported by the National Natural Science Foundation of China [grant numbers 62072199 and 32161133002], and the startup grant of Huazhong University of Science and Technology.
Conflict of Interest: none declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}