





Abstract
Molecular pathway databases represent cellular processes in a structured and standardized way. These databases support the community-wide utilization of pathway information in biological research and the computational analysis of high-throughput biochemical data. Although pathway databases are critical in genomics research, the fast progress of biomedical sciences prevents databases from staying up-to-date. Moreover, the compartmentalization of cellular reactions into defined pathways reflects arbitrary choices that might not always be aligned with the needs of the researcher. Today, no tool exists that allow the easy creation of user-defined pathway representations.
Here we present Padhoc, a pipeline for pathway ad hoc reconstruction. Based on a set of user-provided keywords, Padhoc combines natural language processing, database knowledge extraction, orthology search and powerful graph algorithms to create navigable pathways tailored to the user’s needs. We validate Padhoc with a set of well-established Escherichia coli pathways and demonstrate usability to create not-yet-available pathways in model (human) and non-model (sweet orange) organisms.
Padhoc is freely available at https://github.com/ConesaLab/padhoc.
Supplementary data are available at Bioinformatics online.
1 Introduction
Pathway databases are important bioinformatics tools that support genomics research by providing useful representations of molecular processes. Resources such as KEGG (Kanehisa et al., 2017), Reactome (Croft et al., 2014) or MetaCyc (Karp et al., 2002) offer mechanistic views of metabolic and signaling reactions, whereas other databases such as STRING (Szklarczyk et al., 2017), Omnipath (Türei et al., 2016) or Pazar (Portales-Casamar et al., 2009) feature networks that represent different types of molecular relationships. Pathway databases often use computational resources such as orthology search, text mining and high-throughput assays to create their pathway models, which are usually manually curated by knowledge-domain experts who verify content and add or remove elements prior to public release.
Although the use of pathway databases is widely extended, their ‘create-and-release’ nature implies a number of constraints. First, while cellular processes consist of a virtually unlimited set of interconnected reactions, pathways select and pack them as functional blocks with the topology and boundaries pretty much defined by the curator’s choice. Users are therefore restricted to the provided pathway view, which may not be the best to represent their domain of interest. Second, biological research is highly dynamic, and novel scientific discoveries published in the literature may take years to consolidate in the curated, static pathway databases. This means that investigators at the forefront of research may have difficulties in finding new or updated pathways in established databases, thereby missing the opportunity to use pathway analyses to support their studies.
The scientific community is aware of such limitations, and different solutions have been proposed to offer dynamic pathways that adapt to the needs of the researcher. For example, Reactome (Croft et al., 2014), Biochem4j (Swainston et al., 2017) and LitPathExplorer (Soto et al., 2018) all three allow a versatile interplay between the user and the database content, whereas Wikipathways (Kelder et al., 2012) is one of many community resources that allows for a faster incorporation of novel discoveries (Ostaszewski et al., 2019). Unfortunately, interconnected and community-maintained databases do still fall short to provide tailored biological pathways for new research fields. For example, we failed to find at public pathway databases an integrated representation of the connection between carbon metabolism and epigenetic histone modifications leading to control of gene expression, a research field of growing interest for which a wealth of scientific literature is already available (Cai and Tu, 2011; Mews et al., 2017; Wellen et al., 2009).
Knowledge extraction from scientific literature falls within the domain of text mining. Biomedical text mining has gained ground in recent years, and a large variety of natural language processing (NLP) tools are now available that analyze scientific manuscripts to extract meaningful information. Such tools identify molecular entities, biological relationships and phenotype associations from online papers, and are able to return the information in a structured way (Hirschman et al., 2005; Krallinger et al., 2008). However, there are no tools today that use these technologies to provide a dynamic construction of novel pathways to support the versatility currently required in a fast-evolving research environment and in a user-dependent manner.
In this work, we present Padhoc, a pipeline for Pathway reconstruction ‘on the fly’. Padhoc combines text-mining BioNLP resources with curated databases and Neo4j’s strong visualization capabilities (Miller, 2013) to create novel up-to-date biological pathways tailored to the specific needs of users. We demonstrate Padhoc’s performance on a set of well-known pathways and illustrate its potential for research in model and non-model species by modeling two pathways: the link of metabolism to the epigenetic control of gene expression in Homo sapiens, and the biotic stress response in Citrus sinensis. Padhoc is available at https://github.com/ConesaLab/padhoc.
2 Materials and methods
2.1 Padhoc resources and architecture
2.1.1 Databases and software
Padhoc uses a number of public databases, information resources and functions to construct pathways on the fly. Databases include Brenda (Schomburg, 2004), Omnipath (Türei et al., 2016), IntAct (Kerrien et al., 2012), String (Szklarczyk et al., 2017), Pazar (Portales-Casamar et al., 2009) and ENCODE transcription factor (TF) data (Feingold et al., 2004). PubMed is the source of literature data. Functions and software utilized to extract information from these resources are detailed in Supplementary Table S1.
2.1.2 Text-mining resources
Padhoc makes use of Metrecon (Patumcharoenpol et al., 2016) and TEES (Bj, 2013) as text-mining engines, and of BANNER (Leaman and Gonzalez, 2008) and tmChem (Leaman et al., 2015) as named entity recognition (NER) systems. TEES is a NLP framework developed for the extraction of events and relations from biomedical text. TEES makes use of different training corpora available from the BioNLP Shared Task (Kim et al., 2009) to locate in texts different types of molecular relationships. Metrecon is built on the TEES framework, modifying the training set to extract metabolic reactions. BANNER utilizes a collection of inner dictionaries and is trained with the yeast metabolite corpus (Nobata et al., 2011) to target biological entities across the literature. BANNER has shown high precision, recall and F-Measure scores at the BioCreative Challenge Evaluation (Krallinger et al., 2008). Finally, tmChem is a gold standard for chemical NER (Leaman et al., 2015). Padhoc’s implementation of these NERs facilitates the selection of one or multiple corpora and methods to be used for training, and ensures that all types of biomolecules are efficiently extracted.
2.1.3 Entity ID normalization
In order to provide uniform IDs for genes, metabolites and proteins, names from entities retrieved from public databases are converted into UniProt/ChEBI identifiers using UniProt knowledgebase and tmChem. Names extracted from text mining are more variable in their notations and require additional normalization. Normalization is carried out following NACTEM parsing guidelines (Tsuruoka et al., 2008) that include conversion to lower case, deleting spaces or removing isomer tags among others (see full list in Supplementary Table S2). After normalization, text entities are matched to Neo4j existing nodes using the difflib Python library, and text entities are assigned UniProt/CheBi IDs combining the Neo4j extracted ID with the ID assigned by UniProt knowledgebase or tmChem. In case text-extracted entities cannot be assigned to a UniProt/ChEBI identifier, the BANNER ID is used as Neo4j database node ID.
2.1.4 Homology
Padhoc enables pathway reconstruction for non-model species. When a related model organism is included in the keyword search, the Neo4j database is filled with information from public databases and text extracted from articles of both species. Proteins present in the graph database belong to the queried species and are searched for homology using their UniProt IDs at the InParanoid (O’Brien et al., 2005) web server (http://inparanoid.sbc.su.se/cgi-bin/gene_search.cgi). When orthologous relationships are not present in InParanoid, Padhoc extracts the protein sequences from the UniProt knowledgebase and performs pairwise Blastp similarity searches between the protein sequences from the species. Blastp results with a bit-score >40 are submitted to InParanoid v4.1 for orthology evaluation and eventually incorporated into the Neo4j database. Proteins with inferred orthology relationships are connected in the database using an edge with the label ‘Orthology_relationship’.
2.1.5 Graph compression algorithm
Database elements may still contain redundant information, for example, represent different isomers of the same compound, or two orthologous proteins with the same gene name. In order to further compress data, semantic similarity matrices of protein and metabolite names are constructed using in-house scripts. Briefly, similarities are calculated using difflib after normalizing metabolite and protein names as explained in Section 2.1.3, and clusters of semantically similar features are obtained using the DBSCAN function from scikit-learn (Kramer, 2016; eps = 1.0, min_samples = 1). These clusters are included in the graph database as compressed nodes connected to their corresponding components. Singleton nodes are also included in the compressed graph.
2.1.6 Padhoc installation and utilization
Padhoc is publicly available at https://github.com/ConesaLab/padhoc, and runs in Linux systems with Python v2.7. Padhoc requires the prior installation of TEES, metrecon, Neo4j and tmChem. Guidelines for the installation of these dependencies can be found at Padhoc’s download site. Padhoc is used by running run_padhoc.py script with the list of keywords that represent the pathway to search and the organism of interest. After Padhoc finishes extracting the text and feeding the Neo4j database, the graph is compressed using the script compress_graph.py. After these two steps are completed, the network will be available at the user’s local Neo4j database. More details of how to run Padhoc, as well as the examples used in this article, can be found at Padhoc’s download site.
2.2 Padhoc validation
Padhoc pathways were validated by reconstructing 13 well-established Escherichia coli pathways and comparing them with their annotation in the MetaCyc database (Karp et al., 2002). For this evaluation, Padhoc was directly fed with the scientific literature specified for each pathway in MetaCyc. Entities and relationships from the Padhoc-created pathways that occur or are absent in the reference pathways were treated as true positives and true negatives, respectively, and sensitivity and specificity were calculated. Evaluation was performed filtering entities for increasing levels (from 0 to 5) of literature support, i.e. number of times the entity appears in the literature.
Additionally, Padhoc reconstruction of the E.coli’s Pantothenate and Coenzyme A (CoA) biosynthesis pathway was manually evaluated by human assessment of the relevance of novel Padhoc pathway components and relationships. Features in the Padhoc Pantothenate and Co-A reconstructed pathway were given a relevance score from 1 to 3 where 1 means no relevance, 2 indicates inconclusive relationship and 3 means high relevance for the pathway.
Padhoc was assessed for its ability to reconstruct two de novo pathways. The human histone acetylation pathway was constructed using the keywords ‘Homo sapiens’ and ‘histone acetylation’ and the stress response in citrus species was obtained with keywords ‘biotic stress response’, ‘Citrus sinensis’, ‘Citrus clementina’, ‘Arabidopsis thaliana’ and ‘Physcomitrella patens’. The two last species are model organisms for plants and stress responses, respectively. The novel pathways were evaluated by gene ontology enrichment analysis of recovered genes and by manual comparison to the literature. Additionally, the citrus pathway was evaluated as stress response gene-set in the enrichment analysis of the transcriptional response of citrus to antibiotic treatment (Gardner et al., 2020).
3 Results
3.1 Building a user-defined pathway with Padhoc
Figure 1 shows Padhoc’s scheme for creating a user-defined pathway. Essentially, the procedure starts with a set of keywords that define the pathway the user wishes to create, together with an organism name. The organism name is used to query a compendium of databases (Supplementary Table S1) to retrieve all available metabolic and signaling information for that species, including entity names for genes, metabolites and proteins (and their synonyms), reactions, protein–protein interactions and TF–protein interactions. Molecules are assigned a UniProt and CheBI ID and all information is stored in Neo4j graph database, where nodes represent molecular entities and edges represent the relationships between them (Fig. 1, left).
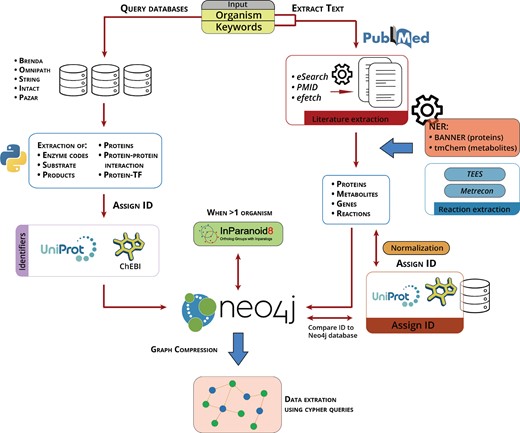
Computational pipeline implemented in Padhoc. The organism name keyword (Left) allows access to different public databases to extract genes, proteins, compounds and molecular relationships. Features are assigned UniProt/ChEBI IDs and incorporated into Neo4j. Organism and pathway keywords (Right) are used to recover relevant literature from Pubmed. Genes, protein, compounds and relationships are extracted using NER resources, text is normalized and incorporated into Neo4j database after assigning a UniProt/ChEBI IDs. InParanoid is used to support homology-based pathway modeling. New pathways are visualized using Neo4j graphical resources
The input set of keywords, together with the organism name, is used to query the scientific literature to retrieve PubMed IDs (PMIDs) and their associated text. Alternatively, a list of PMIDs or text can be supplied as input for Padhoc. TEES and Metrecon programs are then used, in combination with the NER engines BANNER and tmChem, to extract protein names, metabolites and reactions from text. Once the literature data have been extracted, entity names are assigned, when possible, a UniProt/CheBI ID and then added to the Neo4j database. In this process, if a text mining identified entity ID was already present in Neo4j, text mining-derived relationships are incorporated and associated to the existing IDs, otherwise new entity IDs and their relationships are added to the database (Fig. 1, right). Entity names stored in Neo4j are compared with each other and clustered by semantic similarity to create ‘compressed’ nodes that collapse redundant information while maintaining links to source nodes (Supplementary Fig. S1). Finally, when multiple species are submitted to Padhoc (i.e. a non-model organism and a related model species), InParanoid is used to establish orthologous relationships and create hybrid pathways that gather relevant information from both species.
Once the established knowledge (obtained from databases) and emerging knowledge (obtained from text mining) are combined in the Neo4j’s platform, the targeted pathway is retrieved using Cypher queries on the Neo4j database. The resulting biological network can be filtered according to the number of appearances of each entity in the text or can be manually modified by the user. Additionally, customized queries can be used on the newly constructed pathway, e.g. to recover of a set of entities of interest (e.g. a list of genes or metabolites).
3.2 Information content of Padhoc pathways
Padhoc pathway data are stored in a Neo4j graph database and consist of 4 types of nodes and 10 types of relationships. Nodes are either ‘Protein’, ‘Enzyme’ and ‘Compound’, representing the primary structure of the graph (Supplementary Fig. S2A), or ‘Compressed’, which corresponds to a compacted version of semantically similar nodes (Section 2.1.5 and Supplementary Fig. S2B). Every node is assigned a stable ID, which generally corresponds to their UniProt/ChEBI identifier, although some stable IDs use the text name if the entity could not be matched to any database ID. The ten types of relationships include: ‘TM_relationship’, ‘Brenda_database’, ‘StringDB_interaction’, ‘TF_regulation’, ‘Pazar_relationship’, ‘IntAct_interaction’, ‘Omnipath_interaction’, ‘Orthology_relationship’, ‘compressed_to’ and ‘compressed_relationship’. The first eight relationship types define the primary information content of the pathway, whereas the last two are part of the condensed graph.
Pathway links have properties, which store information extracted from text and databases. Hence, Brenda edges contain detailed information of the reaction, the Enzyme Code (EC) of the enzyme that drives the reaction and the species where this reaction was found. Text-mining properties include the training dataset used by TEES (corpora), the type of reaction, the number or appearances in text (nbs), the used search query and the sentences where the reaction was extracted from the text. Omnipath relationships include the post-translational modifications, while the Intact database includes the detection method used to identify interactions, the confidence score, the interaction method and type. In the compressed relationship only sentences are included as a property. Properties stored in each node and relationship types are detailed in Tables 1 and 2.
Node types and properties in the Neo4j database after pathway reconstruction.
| Node label | Properties | Description |
|---|---|---|
| Compound | ID, chebiID, compoundName, textname, sentences and PMID_Tnb | Compound node |
| Protein | Id, uniProtEntryName, uniprotGeneName, uniprotID, uniprotProteinName, species, textname, PMID_Tnb and sentences | Protein node, also enzyme nodes if they catalyze a reaction |
| Enzyme | Id, uniProtEntryName, uniprotID, species, textname, sentences, PMID_Tnb, ECs and synonyms | Enzyme node |
| Compressed | Id, uniprotIDs, Ecs, CompoundNames, chebiIDs and sentences | Compressed nodes and clusters of molecules |
| Node label | Properties | Description |
|---|---|---|
| Compound | ID, chebiID, compoundName, textname, sentences and PMID_Tnb | Compound node |
| Protein | Id, uniProtEntryName, uniprotGeneName, uniprotID, uniprotProteinName, species, textname, PMID_Tnb and sentences | Protein node, also enzyme nodes if they catalyze a reaction |
| Enzyme | Id, uniProtEntryName, uniprotID, species, textname, sentences, PMID_Tnb, ECs and synonyms | Enzyme node |
| Compressed | Id, uniprotIDs, Ecs, CompoundNames, chebiIDs and sentences | Compressed nodes and clusters of molecules |
Node types and properties in the Neo4j database after pathway reconstruction.
| Node label | Properties | Description |
|---|---|---|
| Compound | ID, chebiID, compoundName, textname, sentences and PMID_Tnb | Compound node |
| Protein | Id, uniProtEntryName, uniprotGeneName, uniprotID, uniprotProteinName, species, textname, PMID_Tnb and sentences | Protein node, also enzyme nodes if they catalyze a reaction |
| Enzyme | Id, uniProtEntryName, uniprotID, species, textname, sentences, PMID_Tnb, ECs and synonyms | Enzyme node |
| Compressed | Id, uniprotIDs, Ecs, CompoundNames, chebiIDs and sentences | Compressed nodes and clusters of molecules |
| Node label | Properties | Description |
|---|---|---|
| Compound | ID, chebiID, compoundName, textname, sentences and PMID_Tnb | Compound node |
| Protein | Id, uniProtEntryName, uniprotGeneName, uniprotID, uniprotProteinName, species, textname, PMID_Tnb and sentences | Protein node, also enzyme nodes if they catalyze a reaction |
| Enzyme | Id, uniProtEntryName, uniprotID, species, textname, sentences, PMID_Tnb, ECs and synonyms | Enzyme node |
| Compressed | Id, uniprotIDs, Ecs, CompoundNames, chebiIDs and sentences | Compressed nodes and clusters of molecules |
Type of relationships extracted by Padhoc and stored in Neo4j database for pathway reconstruction
| Label | Properties | Type |
|---|---|---|
| Brenda_database | ECs, reactionsBrenda and species | Enzymatic reactions |
| TM_relationship | Corpora, nbs, query, reactionTypes, sentences and species | Text relationship |
| Omnipath_database | Ptm and species | Signaling |
| PAZAR_database | Metabod, pmid and species | TF-gene regulation |
| STRING_database | No properties | Protein-protein interaction |
| Intact_database | Detection_method, experimental_score, interaction_method, interaction_type and species | Protein-protein interaction |
| Compressed_relationship | Sentences | Condensed graph |
| Compressed_to | No properties | Condensed to uncondensed |
| Orthologous_relationship | No properties | Orthology between proteins |
| Label | Properties | Type |
|---|---|---|
| Brenda_database | ECs, reactionsBrenda and species | Enzymatic reactions |
| TM_relationship | Corpora, nbs, query, reactionTypes, sentences and species | Text relationship |
| Omnipath_database | Ptm and species | Signaling |
| PAZAR_database | Metabod, pmid and species | TF-gene regulation |
| STRING_database | No properties | Protein-protein interaction |
| Intact_database | Detection_method, experimental_score, interaction_method, interaction_type and species | Protein-protein interaction |
| Compressed_relationship | Sentences | Condensed graph |
| Compressed_to | No properties | Condensed to uncondensed |
| Orthologous_relationship | No properties | Orthology between proteins |
Type of relationships extracted by Padhoc and stored in Neo4j database for pathway reconstruction
| Label | Properties | Type |
|---|---|---|
| Brenda_database | ECs, reactionsBrenda and species | Enzymatic reactions |
| TM_relationship | Corpora, nbs, query, reactionTypes, sentences and species | Text relationship |
| Omnipath_database | Ptm and species | Signaling |
| PAZAR_database | Metabod, pmid and species | TF-gene regulation |
| STRING_database | No properties | Protein-protein interaction |
| Intact_database | Detection_method, experimental_score, interaction_method, interaction_type and species | Protein-protein interaction |
| Compressed_relationship | Sentences | Condensed graph |
| Compressed_to | No properties | Condensed to uncondensed |
| Orthologous_relationship | No properties | Orthology between proteins |
| Label | Properties | Type |
|---|---|---|
| Brenda_database | ECs, reactionsBrenda and species | Enzymatic reactions |
| TM_relationship | Corpora, nbs, query, reactionTypes, sentences and species | Text relationship |
| Omnipath_database | Ptm and species | Signaling |
| PAZAR_database | Metabod, pmid and species | TF-gene regulation |
| STRING_database | No properties | Protein-protein interaction |
| Intact_database | Detection_method, experimental_score, interaction_method, interaction_type and species | Protein-protein interaction |
| Compressed_relationship | Sentences | Condensed graph |
| Compressed_to | No properties | Condensed to uncondensed |
| Orthologous_relationship | No properties | Orthology between proteins |
3.3 Padhoc validation using E.coli pathways
Since Padhoc was conceived to create novel pathways, a direct validation of the method is challenging. Therefore, we first evaluated if Padhoc was able to faithfully recapitulate existing pathway data by comparing Padhoc results with curated pathways from established databases. A total of 13 E.coli pathways, representing a wide range of cellular processes of different complexity, were selected from the EcoCyc database (Keseler et al., 2005) for evaluation. The scientific literature reported by EcoCyc as an information source for these pathways, was used as input for our methodology. Supplementary Table S3 lists the 13 biological pathways used for this analysis, as well as the number of relationships that conform each pathway. Figure 2A and B shows the sensitivity and specificity of the method as a function of the supporting evidence (number of appearances in text) for nodes and relationships.
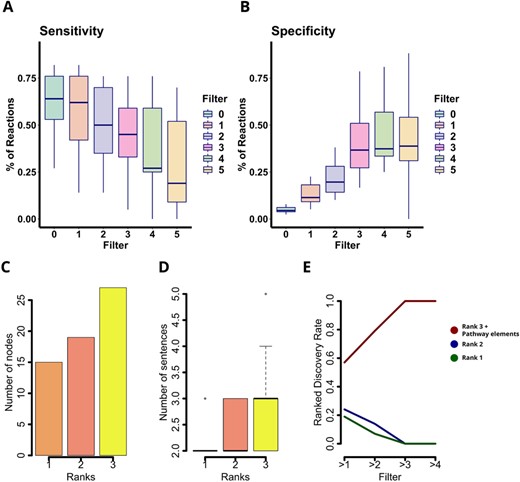
Padhoc evaluation with E.coli pathways. Specificity (A) and sensitivity (B) of reconstructed relationships from E.coli pathways compared with EcoCyc curated database. Manual curation of the Pantothenate and CoA biosynthesis pathway. (C) Number of relationships assigned to each quality rank. (D) Number of sentences supporting each rank assignment. (E) Percentage of confirmed, inconclusive and false pathway calls as a function of the number of supporting sentences
For a support threshold of more than one sentences, Padhoc recovered around 65% of the reactions in the EcoCyc pathways, although only 15% of the reactions in the Padhoc pathways were also present in reference database. For two supporting sentences, mean sensitivity values were 51% and specificity substantially improved to 24% (details for all pathways are provided in Supplementary Table S4). These results suggest that while Padhoc was able to recover most of the elements present in the reference E.coli pathways, a large number of additional entities and links were included when compared with the EcoCyc database. To determine whether new discovered relationships in these pathways were missing pathway information or false additions, Padhoc relationships not present in EcoCyc were manually evaluated and curated for the Pantothenate and CoA biosynthesis pathway. Relationships not present in the EcoCyc pathway were given a score (rank) that represents the quality of the relevance to the pathway. Rank 1 was used for relationships that were recovered from text but either Padhoc did not extract the information correctly or represented descriptions that were not relevant to the pathway (e.g. RNA polymerase reactions when describing molecular biology methods). Relationships were ranked 2 when text was recovered correctly, but their relationship to the pathway was unclear (e.g. folK gene, which interact with 2-amino-6-hydroxymethyl-7,8-dihydropteridine-4-ol as part of tetrahydrofolate biosynthesis pathway). Finally, Rank 3 was given to relationships that extended the EcoCyc pathway in a meaningful way.
Figure 2C shows that over 40% of the added relationships scored 3, indicating that a significant number of novel relationships add relevant knowledge to the pathway. Moreover, Rank 3 sentences show a slightly higher number of appearances in text than relationships with inconclusive relevance for the pathway (Fig. 2D), while sentences with no relevance were barely supported. To identify a suitable support filter, we calculated the rate of bona fide pathway components—i.e. the sum of the rank 3 novel discoveries and the recovered pathway elements when compared with EcoCyc—as a function of the number of occurrences in the text (Fig. 2E). Rank 1 novel discoveries were considered confirmed false positives. As expected, the rate of correctly assigned pathway elements increases with the support and with just more than two supporting sentences reaches 79% of the pathway elements, whereas 14% are inconclusive (Rank 2) and 7% are confirmed false calls (Fig. 2E). Interestingly, > 3 supporting sentences will result in 100% correct assignment of pathway features.
3.4 Reconstruction of human histone acetylation pathways
The ability of Padhoc to reconstruct novel pathways was assessed with the reconstruction of the human histone acetylation pathway, in which metabolic generation of acetyl-CoA supplies the transference of acetyl groups to histone tails through the activity of histone acetyltransferases (HATs). This pathway is gaining scientific interest as it represents an important component of the metabolic control of the epigenetic changes that regulate gene expression. Although several reviews have been already published (Cai and Tu, 2011; Park et al., 2015; Wellen et al., 2009), the information is yet to be included in pathway databases. We used Homo sapiens and ‘histone acetylation’ as keywords for Padhoc and obtained 38 research articles that were used for pathway reconstruction.
Padhoc obtained a network consisting of 562 features (183 genes, 26 metabolites and 353 connections) that were supported by more than one text entry (Supplementary Table S5). The list of the proteins and the metabolites that compose this pathway and their support level is provided in Supplementary Table S6. GO enrichment analysis of the recovered genes returned numerous terms related to histone deacetylation, fatty-acid metabolic process, epigenetic regulation of gene expression, regulation of gene silencing and acetyl-CoA biosynthetic processes among others (Supplementary Table S7), suggesting a successful recovery of gene functions relevant to the targeted pathway. For illustration purposes, we show in Figure 3 the pathway representation after applying a support threshold of three or more sentences, which retains the most relevant elements of this pathway.
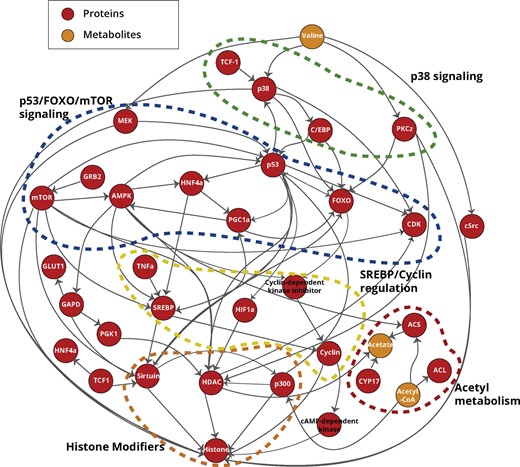
Homo sapiens histone acetylation pathway. Five main subpathways are represented in this network: two signaling pathways (p53, mTOR and FOXO and p38); one transcriptional regulation (SREBP); acetyl-CoA metabolic pathways; and histone modification mechanisms
In agreement with the literature, the pathway recapitulates the connection of acetyl-CoA and acetate with HATs (in this case p300) and histone deacetylases (HDACs, represented by HDAC and Sirtuin), to supply the acetyl group required to acetylate/deacetylate histones, respectively (circled in orange; Wellen and Thompson, 2012; Cai et al., 2011). At central positions of the pathway a highly connected signaling network is observed (circled in blue) including, among other kinases, CDK and mTOR, which are part of the phosphorylation cascade that activates HDACs to regulate their function as histone modifiers (Citro et al., 2015; Masui et al., 2013), and TFs p53 and FOXO, which regulate transcription of growth and apoptotic genes (You and Mak, 2005); p53 is in turn acetylated by p300 (Roy and Tenniswood, 2007) to regulate its activity. The pathway also shows the kinase activation of other TFs controlling the expression of genes involved in nutrient response such as C/EBP (circled in green), regulated by p38 (Wang and Ron, 1996) and SREBP (circled in yellow), controlled by mTOR activity (Shao and Espenshade, 2012). These factors modulate acetyl-CoA availability by regulating the expression of several metabolic genes (circled in red), and are therefore indirect regulators of the acetylation of histones (Shi et al., 2010; Sone et al., 2020). In summary, Padhoc recovered an integrated pathway that joints the metabolic component of HAT- and HDAC-mediated histone acetylation, with the signaling cascade that activates both histone acetylases and the metabolic enzymes linked to acetyl-CoA metabolism.
3.5 Padhoc for pathway reconstruction in non-model species
One of the distinct signatures of Padhoc’s framework is the incorporation of the InParnaoid homology search functionality to facilitate pathway construction for non-model organisms. We evaluated this functionality by using Padhoc to obtain the plant biotic stress response pathway for Citrus sinensis. Citrus are well-studied plants, with available genomic sequences (Wu et al., 2018), where relevant literature on biotic and abiotic stress is available (Martins et al., 2015). However and to our surprise, a molecular map of these stress response mechanisms in citrus—as in other plants—is poorly represented in public databases. The closest pathway in KEGG is the MAPK signaling pathway-plant that describes stress sensing, whereas in PlantCyc (Zhang et al., 2010) there are a total of 498 pathways for Citrus sinensis, but none of them are specifically associated to ‘stress response’. Similarly, the stress response for the plant model species A.thaliana is poorly represented in public databases with the same type of limitations.
Feeding Padhoc with the keywords Citrus sinensis, Citrus clementina, Arabidopsis thaliana, Physcomitrella patens (a fungal model organism) and ‘biotic stress response’, a total of 150 papers were recovered, from which Padhoc extracted 69 genes, 43 metabolites and 111 reactions with more than one supporting sentence (Supplementary Table S8). Genes were enriched in GO terms related to oxidative functions, hormone signaling, response to different stresses, catalytic activities and cellular fluxes. Figure 4 shows the citrus stress response pathway obtained by Padhoc with a filter of two or more supporting sentences. The Neo4j network representation reveals a comprehensive picture of the different molecular events that take place under stress conditions in plants. We observed a cluster of genes and metabolites that represent glutathione metabolism, one of the most important detoxification pathways in plants and animals (Hasanuzzaman et al., 2017). The network also includes numerous metabolic and cellular responses associated to stress. For example, PMD, ADPG genes and pectines are involved in the cell-wall plasticity to adapt to stress conditions (Hamann, 2012); the acetyl-CoA metabolism is known to be down regulated in response to stress (Fatland et al., 2005); Acyl Carrier Protein (ACP) mediates lipid signaling and metabolism under biotic and abiotic stress (Upchurch, 2008); Sucores Phosphate Synthase (SPS) and Starch Synthase (SS) genes are part of the control of sucrose and starch accumulation that follows stress (Julius et al., 2017; Lemoine et al., 2013), while many amino-acids accumulate upon stress conditions to act as osmolytes, regulate ion transport and modulate detoxification (Rai, 2002).
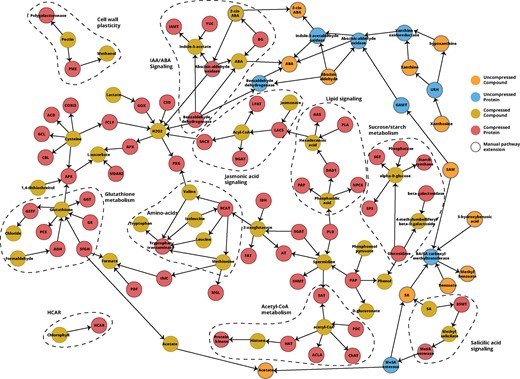
Biotic stress response pathway in C.sinensis obtained by Padhoc. Pathway elements have more than one text supporting sentence
Also important in the Padhoc network is the connection of different plant hormone signaling pathways, including absicic acid (ABA), jasmonic acid and salicylic acid (SA), part of the plant defense signaling systems (Takatsuji and Jiang, 2014), described with different granularity levels both in KEGG and PlantCyc. These hormone pathways are represented in the compressed and filtered Padhoc network by a few members of their signaling cascade. Double clicking any of these compressed nodes allows for the recovery of underlying information available thanks to the inclusion of previous pathway data in the Neo4j database (Fig. 4, orange and blue nodes), revealing new components of the ABA and SA biosynthesis from xanthosine and benzoate, respectively. Node de-compression also revealed that both subpathways are connected by methyltransferases that control enzymatic activity and share S-Adenosyl methionine as substrate. Moreover, the SA pathway is further linked to the glutathione pathway by the utilization of acetate groups as substrate for SFGH esterase (Fig. 4). These results demonstrate the power of Padhoc, not only for joining literature with established knowledge to construct tailored pathways but also in connecting different branches of complex molecular circuits.
Finally, we evaluated this pathway in the context of a gene expression analysis. We used a recent study that evaluated the response of the orange tree to Benzbromarone, a new antibiotics proposed for the treatment of citrus greening (Gardner et al., 2020). Citrus greening is caused by the bacteria Liberibacter asiaticus that affects the floem of the tree leading to dramatic reductions in yields and eventually plant death. In this study, the trunks of affected orange trees were injected with a Benzbromarone infusion to treat infection and leaves were collected after several weeks, to evaluate the transcriptional response of the tree to the antibacterial treatment to understand possible degradation mechanisms for the drug in the plant. The study found 404 genes to be deferentially expressed with respect to mock-treated controls. Enrichment analysis using several pathway resources indicated the activation of sucrose, lignin and cell-wall-related pathways, but no clear insights could were obtained about stress response mechanisms (Gardner et al., 2020). We used the Padhoc citrus stress response pathway as a gene-set for enrichment analysis of the differential expressed genes. We found a significant enrichment for pathway representations at > 1 support (P = 0.014), indicating that a significant stress response was transcriptionally active.
4 Discussion
Biological pathway and molecular interaction databases have evolved as information technology resources to become critical tools in supporting systems biology research, where the assessment of molecular relationships is a fundamental component of the knowledge discovery process. However, the growth of pathway databases is not only sustained by the advance of the computational sciences, but also thanks to the incorporation of a labor- and time-consuming manual curation process that guarantees the quality and completeness of the pathway models. Although this expert review is important to ensure the utility of pathway resources, it has the down-side of resulting in design and coverage restrictions that may limit the application of pathway-based analysis methods to emerging biological domains. Padhoc development was motivated by the realization of the limitations of existing databases for providing adequate data to support pathway analysis in a number of recent studies from our lab.
Padhoc combines knowledge from established pathways databases with a text-mining approach to retrieve the molecular components and interactions for any pathway of interest. This allows the recovery of most state-of-the-art data for biological processes that are under active research, while guaranteeing a curated skeleton of molecular interactions at the base of pathway reconstruction. Padhoc stores the molecular relationships in Neo4j (Miller, 2013), a graph database that offers a user-friendly interface and flexibility to manipulate the pathway. Padhoc’s performance was tested on the reconstruction of a number of E.coli pathways from the EcoCyc database. The assessment of the reconstruction reveals that a large fraction of the recovered pathway features are faithfully recapitulating information from the available literature, but also that Padhoc networks did not capture all the current knowledge on the targeted pathway. This was manifested at E.coli’s Pantothenate and Co-A biosynthesis pathway, where verified calls were nearly 80% of the inferred elements, but 19% of the EcoCyc pathway components were missing. Since both BANNER and tmChem have shown to provide > 90% success in entity recognition from text (Leaman and Gonzalez, 2008; Leaman et al., 2015), this result may indicate that some interactions included in early reference pathways cannot be traced back from the electronically available manuscripts. This limitation should not be as critical for new research domains and is expected to decrease with time, as publication of fully open access manuscripts, amenable to text mining, become a general practice. The manual evaluation of the E.coli Pantothenate and Co-A biosynthesis pathway also revealed that an important amount of bona fide new pathway elements were added by the text mining algorithm. This means that, even for well-established pathway maps, Padhoc was able to provide meaningful updates. At this point of Padhoc development, we have not included in the reconstruction pipeline the reference pathway data, as this would undermine our ability to evaluate the expected performance of Padhoc for newly proposed pathways. However, the flexible structure of the software, where multiple databases are combined and integrated into a unified feature ID system, would make such extensions feasible. Padhoc could also benefit from new NER mechanisms such as HUNER (Weber et al., 2020) and from pathway export into SBML or BioPAX formats, which will be considered in future versions of the software.
To evaluate Padhoc’s capability for constructing pathways not yet available in databases, we used the pipeline to create the histone acetylation pathway in H.sapiens and the biotic stress response in C.sinensis, which also represents analysis scenarios for model and non-model organisms, respectively. In both cases, results showed that Padhoc is able to connect multiple processes that contribute to establish biological functionalities to be modeled as a pathway. Histone acetylation is driven by metabolic processes that lead to acetyl-CoA accumulation and by signaling cascades that control the enzymatic activity of histone modifiers (Wellen and Thompson, 2012; Cai et al., 2011). Although molecular connections that drive histone acetylation can be found at pathway databases, there is no pathway that combines the different mechanisms involved. Padhoc was able to successfully integrate at least five different functional components that contribute to the modification of histones and provided a joint view of this cellular process. Also, in the case of the citrus stress pathway, Padhoc integrated their metabolic, detoxification and hormone signally aspects, generating a comprehensive representation of this response. This is an unique property of our approach, as the definition and number of the keywords used to run Padhoc provide a great deal of flexibility to establish pathway boundaries as a function of the researcher’s needs. Another element of flexibility and versatility of Padhoc is achieved thanks to the utilization of the graphical database Neo4j to host the pathway data. This facilitates, e.g. adjustment of the support threshold to control the confidence and extension of the pathway map. Similarly, the ability to compress/uncompress pathway nodes allows high resolution and navigation on particular aspects of the retrieved network, while uncovering hidden links among its components. This interactive process is particularly useful when using pathway insights for hypothesis generation and mechanistic interpretation of the data.
Finally, a distinctive characteristic of Padhoc is the support for pathway reconstruction also in non-model organisms, thanks to the integration of the InParanoid database and homology search functions. Development of pathway maps for non-model species is usually delayed with respect to model organisms, which imposes a disadvantage to researchers working on these fields. Therefore, the availability of an easy-to-use tool for on-demand pathway construction in these species is particularly useful. We showed Padhoc successfully created a relevant stress pathway in citrus that was effective in providing a suitable gene-set for the analysis of the transcriptional response of the orange tree to antibiotic treatment against citrus greening disease.
5 Conclusions
Overall, Padhoc is an efficient and flexible framework to create pathways ‘on-the-fly’ by extracting relevant information from literature and databases. Padhoc can be used to boost genomics analyses in research fields still poorly represented at established pathway databases. Here, we show Padhoc’s ability to construct biological pathways from the literature, although other applications, such as the compilation of disease maps or the inference of molecular interactions among a set of genes, proteins and metabolites are possible and worth to explore in the future.
Funding
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie [grant agreement no. 675610] and from National Institute of Food and Agriculture, U.S. Department of Agriculture http://nifa.usda.gov/ [award number 2015-70016-23029].
Conflict of Interest: none declared.
References
Sone,H. et al. (2002). Acetyl-Coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am J Physiol Endocrinol Metab, E222-30.
Author notes
The authors wish it to be known that, in their opinion, Salvador Casaní-Galdón and Cecile Pereira should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}