





Abstract
Molecular docking is a widely used technique for large-scale virtual screening of the interactions between small-molecule ligands and their target proteins. However, docking methods often perform poorly for metalloproteins due to additional complexity from the three-way interactions among amino-acid residues, metal ions and ligands. This is a significant problem because zinc proteins alone comprise about 10% of all available protein structures in the protein databank. Here, we developed GM-DockZn that is dedicated for ligand docking to zinc proteins. Unlike the existing docking methods developed specifically for zinc proteins, GM-DockZn samples ligand conformations directly using a geometric grid around the ideal zinc-coordination positions of seven discovered coordination motifs, which were found from the survey of known zinc proteins complexed with a single ligand.
GM-DockZn has the best performance in sampling near-native poses with correct coordination atoms and numbers within the top 50 and top 10 predictions when compared to several state-of-the-art techniques. This is true not only for a non-redundant dataset of zinc proteins but also for a homolog set of different ligand and zinc-coordination systems for the same zinc proteins. Similar superior performance of GM-DockZn for near-native-pose sampling was also observed for docking to apo-structures and cross-docking between different ligand complex structures of the same protein. The highest success rate for sampling nearest near-native poses within top 5 and top 1 was achieved by combining GM-DockZn for conformational sampling with GOLD for ranking. The proposed geometry-based sampling technique will be useful for ligand docking to other metalloproteins.
GM-DockZn is freely available at www.qmclab.com/ for academic users.
Supplementary data are available at Bioinformatics online.
1 Introduction
Zinc is the second most abundant trace transition metal found in living organisms. This is reflected from the fact that about 10% of the structures deposited in protein data bank (PDB: www.rcsb.org) are zinc metalloproteins (Berman, 2000; Burley et al., 2019). Zinc proteins have a multitude of essential functions, including catalysis, storage, transportation, transcription and replication (Anzellotti and Farrell, 2008; Krężel and Maret, 2016; Maret, 2005, 2011, 2012; Parkin, 2004). Central to the functions of these zinc proteins is their zinc-coordination motifs. As shown in Figure 1A, each zinc ion is located at a center coordinated with oxygen (O), nitrogen (N) or sulfur (S) atoms, which can be contributed by either a small-molecule (SM) ligand, a water molecule or an amino-acid residue in the zinc protein. Typical zinc-binding amino-acid residues found in zinc proteins are cysteine, lysine, histidine, aspartate and glutamic acid. Previous analysis (Andreini and Bertini, 2012; Auld, 2001; Maret and Li, 2009) indicates that zinc interacts with the thiolate group in cysteine, the amino group in lysine, one of the two nitrogen atoms of the imidazole ring in histidine, and one oxygen (syn- or anti-) or two oxygen (mono- or bi-) atoms of the carboxylate substituent in glutamate and aspartate whereas the Glu, Asp and water ligands can bridge one zinc ions separately or share several together. Zinc-coordination numbers (CNs) range from 4 to 6 within the first zinc-coordination shell in a tetrahedral, trigonal bipyramidal or octahedral geometry, respectively (Fig. 1B) (Andreini and Bertini, 2012; Auld, 2001; Harding, 2001; Koca et al., 2003; Maret and Li, 2009; Roe and Pang, 1999). Several algorithms have been developed to predict the zinc sites based on the sequences or three-dimensional structures of target proteins (Shu et al., 2008; Zhao et al., 2011). In addition, the CN may be dynamically varied between 4 and 5 or 5 and 6 depending on specific zinc proteins and the atoms in the coordination shells beyond the first shell according to the previous quantum mechanics/molecular mechanics (QM/MM) molecular dynamics (MD) simulations (Dudev and Lim, 2007; Wu et al., 2010).
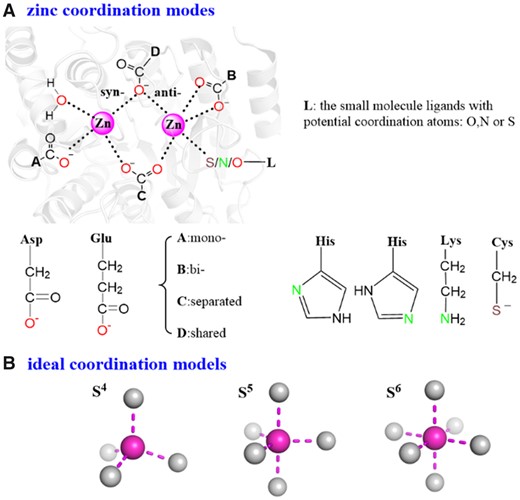
(A) The representative zinc-coordination shell in zinc proteins. The ‘ABCD’ indicate the possible coordination modes of Asp/Glu. (B) The ideal zinc-coordination models (S4, S5 and S6) refer to the standard tetrahedral, trigonal bipyramidal and octahedral geometries
Many zinc proteins are established, or potential, drug targets (Anzellotti and Farrell, 2008; Krężel and Maret, 2016; Parkin, 2004). While progresses were made in molecular docking algorithms (Ballester and Mitchell, 2010; Boyles et al., 2020; Cang and Wei, 2017; Johansson-Akhe et al., 2020; Lu et al., 2019; Schneider et al., 2020; Velazquez-Libera et al., 2020; Wang et al., 2020; Zhang and Sanner, 2019) according to recent assessment (Li et al., 2014; Su et al., 2019), metalloproteins were found more challenging than non-metalloproteins for docking because of additional interactions involving with metal ions. Hu et al. (2004) showed that a correct zinc-coordination geometry is essential for the state-of-the-art docking software FlexX, Autodock and GOLD (Jones et al., 1995, 1997; Kramer et al., 1999; Morris et al., 2009; Rarey et al., 1996; Trott and Olson, 2009) to achieve a reasonable prediction. This leads to several zinc-protein-specific sampling techniques. FlexX (Kramer et al., 1999; Rarey et al., 1996) defines the interaction types and interaction geometry of a metal ion to score protein–ligand interactions in part based on the root-mean-squared deviation (RMSD) between the list of angles in the actual geometry and those in the ideal geometry of the same length. The ideal geometries in FlexX are trigonal bipyramidal, square-based pyramidal, tetrahedral and octahedral. A fragment-based approach is used for ligand docking. Glide XP (Friesner et al., 2004, 2006; Halgren et al., 2004) and GOLD (Jones et al., 1995, 1997), on the other hand, treat metal coordination interactions as special hydrogen bonds. Glide XP performs a grid-based docking conformational search in the functional pocket of the target protein whereas GOLD recognizes both tetrahedral and octahedral geometrical arrangements based on the angles between the metal ion and a pair of coordination positions. Ideal coordination positions in the binding pocket were used to map the ligand acceptors to the coordination positions around the metal ion. AutoDockZn (Santos-Martins et al., 2014) developed a zinc-specific potential to account for both the energetic and geometric (tetrahedral) components of zinc-associated interactions. More recently, force-field-based and knowledge-based scoring functions are combined to improve the ligand-binding ranking for zinc proteins in MpsDockZn (Bai et al., 2015).
In this article, we developed a new zinc-specific method denoted as GM-DockZn for docking a SM ligand onto the zinc-coordination shell of zinc proteins. Unlike previous methods where geometric models were employed as a docking filter, GM-DockZn directly restricts potential coordination atoms in a ligand around ideal geometric models. Moreover, GM-DockZn employed seven ligand-coordination motifs (two in tetrahedral, three in trigonal bipyramidal and two in octahedral geometries) that were found in a survey of zinc protein structures. This new algorithm is shown to significantly improve over several docking programs in locating near-native conformations with correct poses and zinc-coordination motifs within the top 10 or top 50 predictions. Its combination with GOLD yields the highest success rate in top 5 and top 1 predictions.
2 Materials and methods
2.1 Datasets
We obtained 9629 entries of zinc protein structures deposited in the PDB in October 2016. Excluding low-resolution structures (>2.5 Å) led to 6553 zinc proteins with a total of 13 845 zinc-coordination geometries because many proteins contain more than one zinc ion. We further separated these zinc geometries into amino-acid-only (11 589) (AA) and SM containing (2256) structures (see Table 1). Amino-acid-only structures refer to those structures whose coordination positions are all occupied by amino-acid residues. SM-containing structures contain at least one non-amino-acid atom in zinc-coordination positions. These small molecules may be SM ligands or the molecules employed in the solution for crystallization such as water, , PO3− and acetic acid. To avoid the complexity often associated with dynamic solvent molecules, we further extract a single ligand (SL) set (685 structures) from the SM set by limiting the structures with an SL only (no solvent ions) plus amino-acid residues in the first zinc-coordination shell (SL). Here, we defined the first zinc-coordination shell by the distance thresholds: 2.8 Å for Zn–S and 2.5 Å for zinc and the other coordination atoms as before (Andreini and Bertini, 2012; Auld, 2001).
The datasets of zinc proteins (resolution <2.5 Å) from PDB (October 2016) along with the number of structures at different CNs with different coordination motifs (ZN2,2, ZN3,1, ZN2,3, ZN3,2, ZN4,1, ZN3,3 and ZN4,2; ZNP,L, ‘P’ stands for the number of coordination atoms from amino-acid residues and ‘L’ stands for the number of coordination atoms from a ligand)
| Set namea | CN = 4 | CN = 5 | CN = 6 | Total | ||||
|---|---|---|---|---|---|---|---|---|
| AA | 10 057 | 1064 | 468 | 11 589 | ||||
| SM | 1358 | 658 | 240 | 2256 | ||||
| SL | 364 | 244 | 77 | 685 | ||||
| 10 | 354 | 1 | 178 | 65 | 12 | 65 | ||
| ZN2,2 | ZN3,1 | ZN2,3 | ZN3,2 | ZN4,1 | ZN3,3 | ZN4,2 | ||
| Test | 40 | 45 | 23 | 108 | ||||
| NR | 16 | 10 | 6 | 32 | ||||
| HOMO | 20 | 15 | 2 | 37 | ||||
| Set namea | CN = 4 | CN = 5 | CN = 6 | Total | ||||
|---|---|---|---|---|---|---|---|---|
| AA | 10 057 | 1064 | 468 | 11 589 | ||||
| SM | 1358 | 658 | 240 | 2256 | ||||
| SL | 364 | 244 | 77 | 685 | ||||
| 10 | 354 | 1 | 178 | 65 | 12 | 65 | ||
| ZN2,2 | ZN3,1 | ZN2,3 | ZN3,2 | ZN4,1 | ZN3,3 | ZN4,2 | ||
| Test | 40 | 45 | 23 | 108 | ||||
| NR | 16 | 10 | 6 | 32 | ||||
| HOMO | 20 | 15 | 2 | 37 | ||||
AA: all coordination atoms are from amino-acid residues; SM: at least one or more coordination atoms from small molecules; SL: only amino-acid residues and a single ligand (not solvent molecules) contributing to coordination atoms; test: a randomly selected set from the SL set; NR: a NR set at 30% sequence identity cutoff for proteins from the test set; HOMO: 3 proteins with 37 ligand–protein complexes from the test set.
The datasets of zinc proteins (resolution <2.5 Å) from PDB (October 2016) along with the number of structures at different CNs with different coordination motifs (ZN2,2, ZN3,1, ZN2,3, ZN3,2, ZN4,1, ZN3,3 and ZN4,2; ZNP,L, ‘P’ stands for the number of coordination atoms from amino-acid residues and ‘L’ stands for the number of coordination atoms from a ligand)
| Set namea | CN = 4 | CN = 5 | CN = 6 | Total | ||||
|---|---|---|---|---|---|---|---|---|
| AA | 10 057 | 1064 | 468 | 11 589 | ||||
| SM | 1358 | 658 | 240 | 2256 | ||||
| SL | 364 | 244 | 77 | 685 | ||||
| 10 | 354 | 1 | 178 | 65 | 12 | 65 | ||
| ZN2,2 | ZN3,1 | ZN2,3 | ZN3,2 | ZN4,1 | ZN3,3 | ZN4,2 | ||
| Test | 40 | 45 | 23 | 108 | ||||
| NR | 16 | 10 | 6 | 32 | ||||
| HOMO | 20 | 15 | 2 | 37 | ||||
| Set namea | CN = 4 | CN = 5 | CN = 6 | Total | ||||
|---|---|---|---|---|---|---|---|---|
| AA | 10 057 | 1064 | 468 | 11 589 | ||||
| SM | 1358 | 658 | 240 | 2256 | ||||
| SL | 364 | 244 | 77 | 685 | ||||
| 10 | 354 | 1 | 178 | 65 | 12 | 65 | ||
| ZN2,2 | ZN3,1 | ZN2,3 | ZN3,2 | ZN4,1 | ZN3,3 | ZN4,2 | ||
| Test | 40 | 45 | 23 | 108 | ||||
| NR | 16 | 10 | 6 | 32 | ||||
| HOMO | 20 | 15 | 2 | 37 | ||||
AA: all coordination atoms are from amino-acid residues; SM: at least one or more coordination atoms from small molecules; SL: only amino-acid residues and a single ligand (not solvent molecules) contributing to coordination atoms; test: a randomly selected set from the SL set; NR: a NR set at 30% sequence identity cutoff for proteins from the test set; HOMO: 3 proteins with 37 ligand–protein complexes from the test set.
To compare the performance among the multiple methods on an equal footing, 108 zinc proteins were randomly selected from the SL set to serve as the test set (Test). The list of PDB IDs for the Test set along with the details on structural resolution and specific ligands is shown in Supplementary Table S1. This test set has 44 and 26 proteins in common with the test sets for FlexX (Rarey et al., 1996) and MpsDockZn (Bai et al., 2015), respectively. To remove potential biases due to homologous proteins in the test set, we obtained a non-redundant (NR) test set (32 proteins) by excluding proteins with sequence similarity >30% [calculated by CLUSTAL 2.1 (Larkin et al., 2007)] and randomly selecting a representative protein to represent homologous zinc proteins (Supplementary Table S2). We also examine the performance of a method for the same protein (with >90% sequence identity) with different ligands. This set (HOMO) has a total of three representative proteins of 37 ligand–protein complexes with different ligands and CNs (Table 1). The list of PDB IDs for the HOMO set is shown in Supplementary Table S3.
The success rates of locating a correct binding pose within the top 50 predicted poses given by various docking methods for the test set at different CNs
| All (108) (%) | CN = 4 (40) (%) | CN = 5 (45) (%) | CN = 6 (23) (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 35 | 20 | 30 |
| AutoDock4Zn | 43 | 66 | 31 | 30 |
| Glide XP | 44 | 43 | 42 | 48 |
| GOLD | 56 | 53 | 60 | 52 |
| GM-DockZna | 81 | 70 | 87 | 91 |
| All (108) (%) | CN = 4 (40) (%) | CN = 5 (45) (%) | CN = 6 (23) (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 35 | 20 | 30 |
| AutoDock4Zn | 43 | 66 | 31 | 30 |
| Glide XP | 44 | 43 | 42 | 48 |
| GOLD | 56 | 53 | 60 | 52 |
| GM-DockZna | 81 | 70 | 87 | 91 |
This work.
The success rates of locating a correct binding pose within the top 50 predicted poses given by various docking methods for the test set at different CNs
| All (108) (%) | CN = 4 (40) (%) | CN = 5 (45) (%) | CN = 6 (23) (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 35 | 20 | 30 |
| AutoDock4Zn | 43 | 66 | 31 | 30 |
| Glide XP | 44 | 43 | 42 | 48 |
| GOLD | 56 | 53 | 60 | 52 |
| GM-DockZna | 81 | 70 | 87 | 91 |
| All (108) (%) | CN = 4 (40) (%) | CN = 5 (45) (%) | CN = 6 (23) (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 35 | 20 | 30 |
| AutoDock4Zn | 43 | 66 | 31 | 30 |
| Glide XP | 44 | 43 | 42 | 48 |
| GOLD | 56 | 53 | 60 | 52 |
| GM-DockZna | 81 | 70 | 87 | 91 |
This work.
The success rates of locating a correct binding pose within the top 50 predicted poses given by various docking methods for the test set for different Zn-coordination and ligand-chelating motifs
| ZN2,2 (2) (%) | ZN3,1 (38) (%) | ZN2,3 (1) (%) | ZN3,2 (31) (%) | ZN4,1 (13) (%) | ZN3,3 (6) (%) | ZN4,2 (17) (%) | Mono(51) (%) | Bi-(50) (%) | Tri-(7) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| MpsDockZn | 0 | 37 | 100 | 19 | 15 | 33 | 29 | 31 | 22 | 43 |
| AutoDock4Zn | 50 | 63 | 100 | 23 | 54 | 33 | 29 | 61 | 26 | 29 |
| Glide XP | 50 | 42 | 0 | 42 | 46 | 33 | 53 | 43 | 46 | 29 |
| GOLD | 50 | 53 | 100 | 68 | 46 | 67 | 53 | 51 | 62 | 71 |
| GM-DockZna | 50 | 71 | 100 | 94 | 69 | 83 | 94 | 71 | 92 | 86 |
| ZN2,2 (2) (%) | ZN3,1 (38) (%) | ZN2,3 (1) (%) | ZN3,2 (31) (%) | ZN4,1 (13) (%) | ZN3,3 (6) (%) | ZN4,2 (17) (%) | Mono(51) (%) | Bi-(50) (%) | Tri-(7) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| MpsDockZn | 0 | 37 | 100 | 19 | 15 | 33 | 29 | 31 | 22 | 43 |
| AutoDock4Zn | 50 | 63 | 100 | 23 | 54 | 33 | 29 | 61 | 26 | 29 |
| Glide XP | 50 | 42 | 0 | 42 | 46 | 33 | 53 | 43 | 46 | 29 |
| GOLD | 50 | 53 | 100 | 68 | 46 | 67 | 53 | 51 | 62 | 71 |
| GM-DockZna | 50 | 71 | 100 | 94 | 69 | 83 | 94 | 71 | 92 | 86 |
This work.
The success rates of locating a correct binding pose within the top 50 predicted poses given by various docking methods for the test set for different Zn-coordination and ligand-chelating motifs
| ZN2,2 (2) (%) | ZN3,1 (38) (%) | ZN2,3 (1) (%) | ZN3,2 (31) (%) | ZN4,1 (13) (%) | ZN3,3 (6) (%) | ZN4,2 (17) (%) | Mono(51) (%) | Bi-(50) (%) | Tri-(7) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| MpsDockZn | 0 | 37 | 100 | 19 | 15 | 33 | 29 | 31 | 22 | 43 |
| AutoDock4Zn | 50 | 63 | 100 | 23 | 54 | 33 | 29 | 61 | 26 | 29 |
| Glide XP | 50 | 42 | 0 | 42 | 46 | 33 | 53 | 43 | 46 | 29 |
| GOLD | 50 | 53 | 100 | 68 | 46 | 67 | 53 | 51 | 62 | 71 |
| GM-DockZna | 50 | 71 | 100 | 94 | 69 | 83 | 94 | 71 | 92 | 86 |
| ZN2,2 (2) (%) | ZN3,1 (38) (%) | ZN2,3 (1) (%) | ZN3,2 (31) (%) | ZN4,1 (13) (%) | ZN3,3 (6) (%) | ZN4,2 (17) (%) | Mono(51) (%) | Bi-(50) (%) | Tri-(7) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| MpsDockZn | 0 | 37 | 100 | 19 | 15 | 33 | 29 | 31 | 22 | 43 |
| AutoDock4Zn | 50 | 63 | 100 | 23 | 54 | 33 | 29 | 61 | 26 | 29 |
| Glide XP | 50 | 42 | 0 | 42 | 46 | 33 | 53 | 43 | 46 | 29 |
| GOLD | 50 | 53 | 100 | 68 | 46 | 67 | 53 | 51 | 62 | 71 |
| GM-DockZna | 50 | 71 | 100 | 94 | 69 | 83 | 94 | 71 | 92 | 86 |
This work.
2.2 The deviation from ideal orientations: RMSDOR
We employ RMSDOR to measure the orientational RMSDs of observed geometries from the ideal geometries of tetrahedral, trigonal bipyramidal and octahedral models. It is defined as the minimum RMSD between the coordinates of unit vectors along the direction of Zn to a coordination atom in the observed structure and those in the ideal model. This definition allows us to focus on the orientational deviations by ignoring atomic distance fluctuations (e.g. the bond length Zn–S is longer than zinc to other atoms). This is similar to the work by Seebeck et al. (2007) who calculated RMSD based on the angles between the vectors from the zinc to a coordination atom.
2.3 Zn-ligand coordination pose prediction
The schematic diagram for ligand docking is shown in Figure 2. Before docking, all ligands and solvent molecules in the query PDB structure are removed. Then, the following procedure is employed. The first step is to locate all current coordination atoms in the query PDB structure according to the distance criterion and align these coordination atoms with all possible ideal models by minimizing RMSDOR. The locations of any missing coordination atoms in an ideal model with RMSDOR less than a cutoff value (0.25 Å) are considered as the potential locations of ligand coordination atoms.

The flow chart of the ligand-docking protocol of the GM-DockZn in this work. Step 1: Identify potential coordinating atom(s) in a ligand and locate their potential positions according to RMSDOR from ideal models (only three examples were shown); step 2: determine the initial ligand position; step 3: place ligand onto the target protein by rotation and translation to generate various binding poses; step 4: exclude poses with steric hinderance from amino-acid residues and the zinc center and step 5: rank poses by evaluating the zinc-ligand binding affinity based on the Amber99sb-SLEF force field
According to our statistical analysis of the SM set, the number of missing coordination atoms contributed by an SL can be between 1 and 3. Thus, we treat these three cases sequentially. We start with the case that the ligand provides a single atom for coordination. In this case, the possible positions of the ligand’s coordination atom a1 can be generated by using a grid of 0.1 Å between a distance of 2 and 2.4 Å (or to 2.8 Å for S atom) and a step of 30° for angles (θ,φ) on the spherical surface centered at the zinc and around the ideal position (Fig. 2). Only those positions with RMSDOR < 0.25 Å are kept. Then, all O, N and S atoms in a ligand are considered as atom a1 in turn as a potential coordination atom. Afterwards, possible coordinates of the nearest-connecting atom a2 are obtained by taking a1 as the center, the distance of a1–a2 as the radius and 30° for angles (θ,φ) on the spherical surface for sampling. Only those positions with their distances to zinc (r(Zn, a2)) > 2.5 Å are kept for a2. The positions of the third atom a3 are sampled by taking a1–a2 as the rotation axis with a 30° interval at a fixed angle a1–a2–a3 and the distance between a2 and a3. Only those positions with r(Zn, a3) > 2.5 Å are kept for a3. Once the positions of a1, a2 and a3 are known, the whole ligand pose can be obtained by building on the xyz-coordinates of the a1, a2 and a3 atoms and the internal coordinate system of the ligand. Next, we examine the case that a ligand provides two atoms as coordination atoms. The possible positions of a1 are obtained as in the case of a single-coordination atom from the ligand. The possible positions of a2 are sampled by a 30° interval with the vector Zn–a1 as the rotation axis at the fixed ideal distance between a1 and a2 and the fixed ideal angle of Zn–a1–a2. Only those a1 and a2 positions with RMSDOR < 0.25 Å are kept. Once the positions of a1 and a2 are known, the third atomic positions and the entire ligand can be built as described before. Finally, in the case of a ligand providing three atoms as coordination atoms, the first two atoms (a1 and a2) are done as before. Then, the possible positions of a3 are sampled in a 30° interval with a1–a2 as the rotation axis and the fixed angle a1–a2–a3 and the distance between a2 and a3. Only those positions with 1.9 Å < r(Zn, a3) < 2.5 Å are kept for a3. For all possible three atomic positions as coordination atoms, only those with RMSDOR < 0.25 Å from ideal models are kept. Once three atomic positions are known, the conformation of the whole ligand can be obtained as before. To avoid steric clashes, all above-generated ligand poses are removed if any atoms of the ligand are within 2 Å from any heavy atoms of the protein (cutoff) or 2.5 Å from zinc.
Here, the threshold of RMSDOR (<0.25 Å) and the conformational step size of ligand (0.1 Å and 30°) are user-defined parameters. These default values employed herein are recommended to balance the efficiency and accuracy of docking.
2.4 Poses scoring
All van der Waals parameters and partial charges for zinc interactions in Equations (1)–(4) are obtained from the Amber99sb-SLEF force field. α, β, λ and R* parameters in (3) and (4) optimized by QM/MM force are 0.11 (Å2/e2), 0.81 (Å−2), 0.74 (Å−1) and 1.36 (Å), respectively (Gong et al., 2015).
2.5 Other methods
GOLD version 4.1.2 was used for the redock experiments. Glide XP module was from Schrödinger (Schrödinger, LLC: New York, NY, 2015). AutoDock4Zn was downloaded from its official website: autodock.scripps.edu. MpsDockZn was kindly provided by Dr. Honglin Li.
2.6 Docking preparation
All apo-protein structures are generated by using the Molecule Operating Environment package (MOE, 2013) to remove ligands from the native proteins. The hydrogen atoms of proteins are also added and pre-optimized by the MOE software suite. The general amber force field (Wang et al., 2004) was applied for all SM ligands and their atomic charges were assigned from restrained electrostatic potential calculations at the HF/6-31G* theoretical level in Gaussian 09 package (Frisch, 2009).
2.7 Performance measure
A ligand pose is evaluated according to the RMSD of the structure superposition between a predicted pose and the native structure based on all heavy atoms of the ligand in the presence of fixed zinc and protein positions. In addition to RMSD, docking performance is also measured by the reproducibility of the correct CN and the coordination atom in the native crystal structure because low RMSD conformations may be associated with an incorrect zinc-coordination structure. Here, a successful redocking is defined as RMSD < 2.0 Å with the correct CN and coordination atoms. A RMSD cutoff value of 2.0 Å was also used previously for evaluation (Bai et al., 2015; Santos-Martins et al., 2014).
3 Results
3.1 RMSDOR distribution
To choose a cutoff for the deviation of a ligand-containing coordination system from ideal models, one needs to know the natural fluctuation around the ideal models in zinc proteins complexed with small molecules. Figure 3A shows the distribution of RMSDOR with 4, 5 and 6 CNs, respectively, in the SM set that has 2256 zinc proteins complexed with one or more small molecules. The results show that over 95% of 2256 zinc-coordination systems have an RMSDOR value lower than 0.25 Å. As a result, RMSDOR of 0.25 Å is used as a default cutoff to remove those structures away from the ideal coordination models. Interestingly, the distribution of RMSDOR for 5-coordination systems is quite similar to those obtained from QM/MM MD calculations on ligand binding of HDAC (see Supplementary Fig. S1).
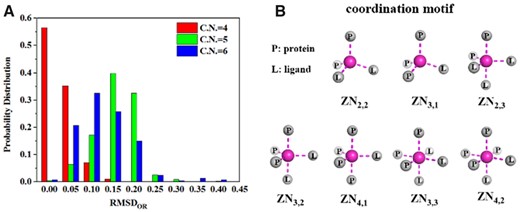
(A) The distribution of the geometry matching parameter (RMSDOR) generated from the SM set. The majority (99.9% of 4-, 96.4% of 5- and 95.1% of 6-coordination zinc structures) have an RMSDOR value of lower than 0.25 Å. (B) All possible coordination motifs (seven) found in the PDB structures of the SL set, denoted by ZNP,L with P, L are the number of atoms from proteins and a ligand, respectively
3.2 Possible coordination motifs
We examined the possible zinc-coordination motifs in the presence of an SL. Using the SL set (685 protein–ligand complexes), we found that there are only 7-coordination motifs (Fig. 3B), which are annotated by ZNP,L with P, L are the number of atoms from proteins and a ligand, respectively. The occurrences of these motifs are listed in Table 1. An SL in the tetrahedral geometry can contribute one (354 structures) or two coordination atoms (10 structures). An SL in the trigonal bipyramidal geometry can contribute one on the triangle plane (65 structures), one on and one off the triangle plane (178 structures) and two on and one off the triangle plane (1 structure). An SL in the octahedral geometry can contribute two (65 structures) or three (12 structures) coordination atoms.
3.3 Docking results
We examine the performance of GM-DockZn by using a test set of 108 protein–ligand complexes. To make a comparison to other methods on an equal footing as much as we can, we obtain 50 top-ranked poses from all methods compared. Because the default output number of GOLD and AutoDockZn is <10, we modified the minimum-energy cutoff so that we can obtain at least 100 docking poses to facilitate comparison. The resulting docking time for GOLD and AutoDockZn is about 10 times longer than the default.
Figure 4A summarizes the performance on the test set by five methods. The performance is measured by plotting the best RMSD value among 50 best poses predicted for each target from small to large. The lower the curve is, the better the performance is. For the first few best predictions, all methods have similar performance in term of RMSD with GOLD having a slight edge. However, all methods except GM-DockZn made false-positive predictions that have small RMSD values but with either incorrectly predicted zinc-coordination structures, or incorrect coordination atoms in the ligand, or both (shown as open circles). Moreover, GM-DockZn has the highest number of the poses with RMSD ≤ 2 Å [88/108 versus 67/108 by the next best (GOLD) and 60/108 after excluding those with incorrectly predicted coordination atoms or numbers, Table 2]. That is, GM-DockZn makes 47% (88/60) increase in success rate over the second-best GOLD in reproducing the correct binding pose and CN around the zinc ion.
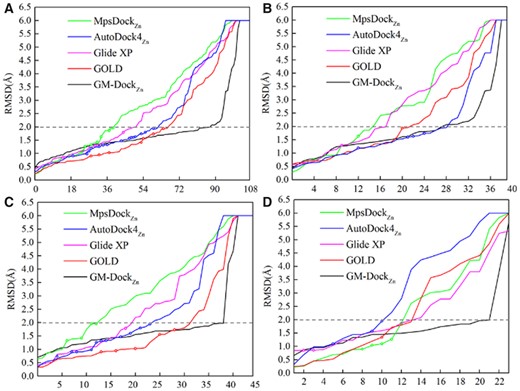
(A) Method performance given by five docking algorithms as labeled according to the best binding pose (in RMSD, the y-axis) in the top 50 predictions for each target arranged in the increasing order (number of targets in the x-axis) for the whole test set. (B), (C) and (D): Same as (A) but the performance for 4-, 5- and 6-coordination systems, respectively. An RMSD cutoff of 2.0 Å is shown as a dashed line and >6.0 Å for truncation. Close and open circles are true and false-positive predictions, respectively. False-positive predictions are those predictions with RMSD ≤2.0 Å but incorrectly predicted CNs or coordination atoms
Figure 4B–D compares the performance for 4-, 5- and 6-coordination systems, respectively. For the 4-coordination system, AutoDock4Zn has essentially the same performance as GM-DockZn in term of the number of the poses with RMSD ≤ 2 Å. Both have a much higher number of correctly predicted ligand binding poses than all other methods. However, AutoDock4Zn made several false-positive predictions. For 5- and 6-coordination systems, GOLD is the second best although it contains false-positive predictions as well. GM-DockZn is the only one having the highest number of binding poses with RMSD ≤ 2 Å in the absence of any false positives. It is noted that there is a sudden increase RMSD at RMSD > 2 Å for 5- and 6-coordination systems. This is largely due to the finite grids we used in GM-DockZn to map possible binding poses. If a near-native binding pose is not found, the next near-native binding pose will have a significantly different structure.
Table 2 summarizes the success rate of locating a correct pose within the top 50 predicted poses (specific results for each structure by all methods are shown in Supplementary Table S4). This result is obtained after removing false-positive predictions by other methods. GM-DockZn makes an absolute improvement over the next best AutoDock4Zn by 4% in the 4-coordination system, 27% over the next best GOLD in the 5-coordination system and 39% over the next best GOLD in the 6-coordination system. GM-DockZn achieves 0% false-positive rates, compared to 10% by GOLD, 26% by AutoDock4Zn, 8% by Glide XP and 29% by MpsDockZn.
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by various methods for the test set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 9 | 5 | 2 |
| AutoDock4Zn | 43 | 27 | 20 | 17 |
| Glide XP | 44 | 34 | 17 | 14 |
| GOLD | 56 | 47 | 35 | 26 |
| GM-DockZn | 81 | 67 | 34 | 17 |
| GM-DockZn + GOLD | 72 | 53 | 47 | 31 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 9 | 5 | 2 |
| AutoDock4Zn | 43 | 27 | 20 | 17 |
| Glide XP | 44 | 34 | 17 | 14 |
| GOLD | 56 | 47 | 35 | 26 |
| GM-DockZn | 81 | 67 | 34 | 17 |
| GM-DockZn + GOLD | 72 | 53 | 47 | 31 |
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by various methods for the test set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 9 | 5 | 2 |
| AutoDock4Zn | 43 | 27 | 20 | 17 |
| Glide XP | 44 | 34 | 17 | 14 |
| GOLD | 56 | 47 | 35 | 26 |
| GM-DockZn | 81 | 67 | 34 | 17 |
| GM-DockZn + GOLD | 72 | 53 | 47 | 31 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| MpsDockZn | 28 | 9 | 5 | 2 |
| AutoDock4Zn | 43 | 27 | 20 | 17 |
| Glide XP | 44 | 34 | 17 | 14 |
| GOLD | 56 | 47 | 35 | 26 |
| GM-DockZn | 81 | 67 | 34 | 17 |
| GM-DockZn + GOLD | 72 | 53 | 47 | 31 |
Table 3 further displays the performance of different methods for seven possible coordination geometries along with mono-, bi- and tri-chelating ligands to the zinc. Except for some geometries with few cases (2 for ZN2,2 and 1 for ZN2,3), GM-DockZn makes consistent improvement in all other geometries and all chelating possibilities. This indicates the robustness of performance improvement.
The above comparisons were based on the top 50 predictions. Table 4 further compares the success rate for the top 50, top 10, top 5 and top 1 predictions. GM-DockZn improves over the second-best GOLD significantly at the top 50 and 10 predictions but is only comparable for the top 5 and worse for the top 1 prediction. This indicates that GM-DockZn achieves the best in sampling but the force field employed in this work is not the best for ranking.
To further improve the usefulness of GM-DockZn for docking, we examine the possibility of using GM-DockZn for sampling and GOLD for scoring. The results of the combined method, which is labeled as GM-DockZn + GOLD, are shown in Table 4. We found that GM-DockZn + GOLD substantially improves over GOLD and GM-DockZn in top 5 (>12%) and top 1 prediction (>5%) in term of the success rate for sampling near-native conformations. The results confirm the power of using a better scoring function in ranking the poses obtained by GM-DockZn.
In the above method comparison for zinc-binding proteins (Cinaroglu and Timucin, 2019; Santos-Martins et al., 2014), homologous proteins are often not excluded because even the same protein may bind different ligands differently in term of their poses, CNs and coordination atoms. Nevertheless, it is necessary to examine the effect of binding to different proteins and binding to the same protein, separately. We have made NR and homolog sets for this purpose (see Section 2). As shown in Figure 5, GM-DockZn continues to have the best performance for both NR (Fig. 5A and Supplementary Table S5) and HOMO (Fig. 5B and Supplementary Table S6) sets. However, the improvement of success rate is smaller for the NR set (13% absolute improvement over the next best GOLD, compared to 25% in the whole test set) and larger for the HOMO set (30%). This indicates that NR provides a more realistic estimation of improvement without homology biases. On the other hand, the results on the HOMO dataset indicate that GM-DockZn is more capable of handling different ligands docking into the same structure.
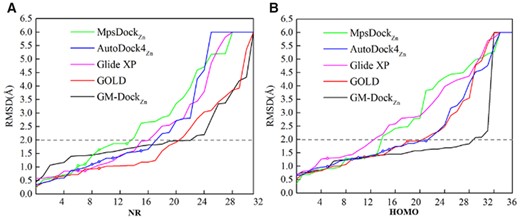
Same as Figure 4 but for the performance on (A) the NR set (NR) and (B) the homology set (HOMO), respectively
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by three methods for the set of 20 apo-proteins in the NR test set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 30 | 25 | 20 | 10 |
| GM-DockZn | 45 | 30 | 20 | 10 |
| GM-DockZn + GOLD | 50 | 30 | 20 | 15 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 30 | 25 | 20 | 10 |
| GM-DockZn | 45 | 30 | 20 | 10 |
| GM-DockZn + GOLD | 50 | 30 | 20 | 15 |
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by three methods for the set of 20 apo-proteins in the NR test set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 30 | 25 | 20 | 10 |
| GM-DockZn | 45 | 30 | 20 | 10 |
| GM-DockZn + GOLD | 50 | 30 | 20 | 15 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 30 | 25 | 20 | 10 |
| GM-DockZn | 45 | 30 | 20 | 10 |
| GM-DockZn + GOLD | 50 | 30 | 20 | 15 |
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by three methods for all possible combinations of 452 cross-docking results in the HOMO set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 61 | 46 | 36 | 13 |
| GM-DockZn | 76 | 48 | 37 | 14 |
| GM-DockZn + GOLD | 70 | 63 | 58 | 35 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 61 | 46 | 36 | 13 |
| GM-DockZn | 76 | 48 | 37 | 14 |
| GM-DockZn + GOLD | 70 | 63 | 58 | 35 |
The success rates of locating a correct binding pose within the top 50, top 10, top 5 and top 1 predicted poses given by three methods for all possible combinations of 452 cross-docking results in the HOMO set
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 61 | 46 | 36 | 13 |
| GM-DockZn | 76 | 48 | 37 | 14 |
| GM-DockZn + GOLD | 70 | 63 | 58 | 35 |
| Top 50 (%) | Top 10 (%) | Top 5 (%) | Top 1 (%) | |
|---|---|---|---|---|
| GOLD | 61 | 46 | 36 | 13 |
| GM-DockZn | 76 | 48 | 37 | 14 |
| GM-DockZn + GOLD | 70 | 63 | 58 | 35 |
The above results were obtained by docking onto holo-structures. To investigate the effect of protein conformational changes on docking performance, we employed 20 proteins from the NR set with apo-structures. The list of PDB IDs is shown in Supplementary Table S7. As shown in Table 5, GM-DockZn continues to have the best performance for obtaining near-native structures within top 50 and top 10. Interestingly, GM-DockZn and GOLD have comparable performance for apo-docking. GM-DockZn + GOLD, further improves over GM-DockZn and GOLD in top 1 and top 50.
Another way to examine the effect of conformational transitions is to perform cross-docking between different protein–ligand complexes of the same protein. We performed all possible combinations of cross-docking of the structures for the same protein in the HOMO set (listed in Supplementary Table S3). The corresponding results are summarized in Table 6. GM-DockZn continues to have a significant improvement in top 50 over GOLD. It also shows a slightly better performance than GOLD for top 10, top 5 and top 1. Improved top 1 performance relative to GOLD by GM-DockZn suggests that GM-DockZn is less affected by small conformational changes. The combination of two methods (GM-DockZn + GOLD), although slightly worse than GM-DockZn in top 50, makes a significant improvement in top 10, top 5 and top 1 (15%, 21% and 21% absolute improvement in success rate).
We illustrate the difference between GM-DockZn and other methods in more details by using an example for CNs of 4, 5, 5 and 6 (Fig. 6). The best poses in the top 50 for six methods are shown. For the coordination of four atoms around zinc, the crystal structure of thermolysin (TLN, PDB code: 1ZDP) (Bradner et al., 2010) was used as an illustration. Zinc is surrounded by three coordinative atoms from two HIS and one GLU residues and one sulfur atom from the ligand (2-mercaptomethyl-3-phenyl-propionyl-glycine). The best pose from Glide XP has a large RMSD of 3.6 Å with an incorrect coordination atom from the ligand (O instead of S). All other methods (GM-DockZn, GOLD, AutoDock4Zn and MpsDockZn) provide a reasonable prediction with the correct coordination atom and an RMSD value of <2 Å.
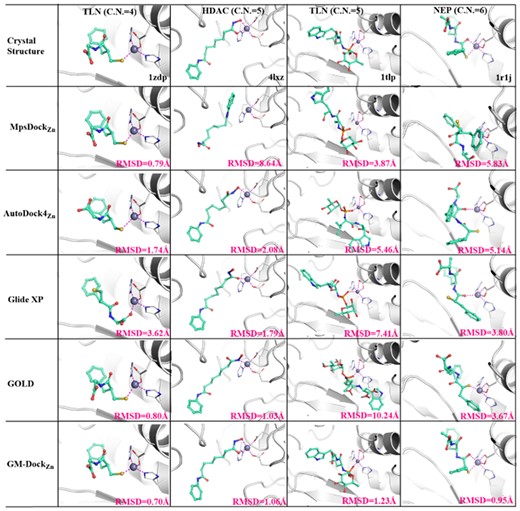
The illustrative examples of redocking results for four zinc-coordination structures with the CN of 4, 5, 5 and 6, respectively. The best pose in the top 50 for each method is shown
For the CN of 5, two typical examples are shown with HDAC2 (4LXZ) (Lauffer et al., 2013) and TLN (1TLP) (Tronrud et al., 1986). In HDAC2, the zinc-coordination shell is a square-pyramid geometry, with two ASP and one HIS residues providing three coordinative atoms while the ligand SAHA is bi-chelating to the zinc ion. For this example, GM-DockZn, is the only one successful to generate a near-native pose. As shown in Figure 6, Glide XP, GOLD and AutoDock4Zn failed to produce the bidentate feature of SAHA. That is, they can only predict a monodentate coordination mode. MpsDockZn is unable to rank the coordination between zinc and SAHA within the top 50. For TLN (1TLP), the zinc-coordination shell is a trigonal bipyramidal geometry, made of one bidentate GLU, two HIS residues and a mono-chelating ligand. Similarly, GM-DockZn is also the only method that correctly reproduces the native zinc-coordination shell in the crystal structure. It should be noted that the RMSD values are reasonable for GOLD and Glide XP in HDAC2 but coordination details are incorrect.
For the 6-atom zinc-coordination shell, the neprilysin protein bound with its inhibitor ORI [n-(3-phenyl-2-sulfanylpropanoyl) phenylalanylalanine] is selected as a representative case (Oefner et al., 2004). The octahedral coordinative geometry consists of two His and one bidentate Glu residues, as well as a bidentate ligand (ORI). As shown in Figure 6, the CN is only 5 for the best poses given by GOLD, Glide XP and AutoDock4Zn, with the ligand in a monodentate pose instead of bidentate. There is no chelation interaction between the ligand and the zinc ion according to MpsDockZn. GM-DockZn is the only method that successfully yields the correct binding mode and coordination motif.
4 Discussion
In this paper, we have developed a geometry-based docking technique for zinc proteins. In this technique, all potential coordination atoms in a ligand are placed near-ideal locations of seven discovered ligand-coordination motifs by a grid search. The method provides a substantially improved capability over several methods in sampling near-native poses that are not only low in RMSD but also correct in term of CNs and coordination atoms. Many existing methods can yield low RMSD poses but with incorrectly predicted coordination atoms or CNs. The results highlight the importance of using more than RMSD in docking assessment for metalloproteins.
Our systematic investigation of coordination motifs only found seven possible geometries as shown in Figure 3B. That is, not all possible combinations of ligand positions are present. This may be interpreted by the requirement that the interactions between zinc and protein atoms and between zinc and ligand atoms have to be both strong. For example, a coordination motif in the tetrahedral geometry with three coordination atoms from a ligand will lead to only a single atom in the protein to interact with zinc. This interaction will likely be too weak for a protein to retain zinc and thus this coordination motif does not exist. On the other hand, two or three ligand atoms are needed in the octahedral geometry to retain the ligand. Otherwise, the zinc will be overwhelmed by interaction with five protein atoms.
GM-DockZn makes a grid search on a spherical surface. In principle, we could further improve conformational sampling with finer grids. However, this would be at a significant cost in the computing time. With the current parameter sets for sampling top 50 poses, it takes about 16 CPU h for GM-DockZn to complete the NR set on a single CPU of a personal computer (Intel Xeon E3-1240V5 Quad core 3.5 GHz), compared to about 12 h by GOLD, 16 h by AutodockZn, 20 h by Glide XP and 28 h by MpsDockZn on the same single CPU. Nevertheless, we have examined the effect of using different angle grids (10, 15, 20, 30 and 45) on the success rates of locating near-native poses by using the NR set. As shown in Supplementary Table S8, we confirmed an improvement in the top 50 poses sampled at 20° and 15° grids but with 3 and 8 times increases in computing times, respectively. However, a further reduction to 10° did not improve the sampling. This is mainly because more conformations increase the challenge for the scoring function to recognize the near-native structures. This is also reflected from the variation of the best grid for top 50, top 10, top 5 and top 1 (15°, 30°, 20° and 15°, respectively). The results highlight the critical need for improving the scoring function.
GM-DockZn employs the AMBER force field (ff99SB) for protein–ligand interactions and the SLEF force field for the zinc-protein and zinc-ligand interactions. Although the entropic effect and the solvation free energy were not considered, the combination of the force fields has allowed a reasonable selection of the top 50 predictions. To examine the effect of the SLEF force field, we have reperformed scoring of the NR set in its absence. As shown in supplementary Table S9, we found that including SLEF leads to 7% and 11% absolute increase in the success rate for sampling near-native conformations for top 50 and top 10 conformations, respectively, but 8% and 2% decreases for top 5 and top 1 conformations, respectively. This suggests that further improvement of the SLEF force field is required for more accurate detection of near-native conformations.
The current implementation of GM-DockZn has been focused on conformational sampling. We expect that GM-DockZn could be further extended for other metalloproteins, if there is a sufficient number of known protein structures for characterization of the coordination motifs for other metalloproteins. For example, we can directly use the 6-coordination model for most magnesium metalloproteins but possible motifs with ligand atomic positions would require statistical analysis of their existing structures. Calcium ions, on the other hand, have a representative 8-coordination system and thus an 8-coordination model should be employed by a survey of typical protein complexes to characterize the calcium binding motifs. Once binding motifs with known positions of possible ligand atoms are resolved, it is straightforward to employ the approach developed here.
One weakness of the current implementation is that it does not sample all possible rotatable bonds in ligands. Another weakness is that it is unable to locate the best within the top 5 or top 1 as shown in Table 4. Despite of the weakness, we found that the performance of GM-DockZn is more robust when docking onto apo-structures and when cross-docking between the structures of the same protein complexed with different ligands (Tables 5 and 6). To make GM-DockZn more practically useful, we have developed a combined method GM-DockZn + GOLD: using the GOLD scoring function to score the conformations sampled by GM-DockZn. Results on docking onto holo-structures, apo-structures and cross-docking consistently showed that GM-DockZn + GOLD provides a significant improvement in detecting near-native poses within top 1 and top 5 by combining near-native sampling by GM-DockZn with accurate ranking capability of GOLD. We expect a similar superior performance of GM-DockZn + GOLD for the structures from homology modeling because the grid-based sampling quality of GM-DockZn is less sensitive to small conformational transitions near the metal active site. Including rotatable bonds in GM-DockZn is working in progress.
The above usage of GOLD for scoring GM-DockZn provides a practical solution to this challenging problem of metalloprotein docking. To go beyond GM-DockZn + GOLD, it is necessary to develop a next-generation scoring function with improved characterization of metal–ligand and metal–protein interactions. In addition to traditional empirical and quantum-mechanical-based force fields employed here, machine-learning plays an increasingly important roles in docking scoring (Nguyen and Wei, 2019; Nguyen et al., 2020). A machine-learning-based scoring function will be likely useful for ligand docking on metalloproteins as more and more structural data become available.
Acknowledgements
We thank the Guangzhou and Shenzhen Supercomputer Center for providing computational source. And we also thanks for the Three Big Constructions-Supercomputing Application Cultivation Projects from SYSU. The day of resubmission is also the 6th birthday of my (Ruibo Wu) son (Zhengyu Joey Wu), who gave me many inspirations on science, herein I want to wish him a happy childhood.
Funding
This work was supported by the National Natural Science Foundation of China (21773313, 21803079), the National Key R&D Program of China (2017YFB0202600) and Shenzhen Science and Technology Program (grant no. KQTD20170330155106581).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, KaiWang and Nan Lyu should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}