





Abstract
Simulated sequence alignments are frequently used to test bioinformatics tools, but current sequence simulators are limited to defined state spaces. Here, we present the COMPletely Arbitrary Sequence Simulator (COMPASS), which is able to simulate the evolution of absolutely any discrete state space along a tree, for any form of time-reversible model.
COMPASS is implemented in Python 2.7, and is freely available for all platforms with the Supplementary Information, as well as at http://labs.carleton.ca/eme/software-and-data.
Supplementary data are available at Bioinformatics online.
1 Introduction
Simulated data are often used to assess the performance of new bioinformatic algorithms, test the behavior of biological models, and to generate null distributions through parametric bootstrapping. For example, multiple sequence alignments simulated under known parameter values are used to test the accuracy and robustness of methods for statistical inference from phylogenetic data (Lartillot et al., 2009; Yang and Nielsen, 2000). There are a number of packages available for the simulation of sequence alignments, but they tend to operate over a defined state space and/or implement specific evolutionary models. For example, INDELible offers substantial flexibility with respect to models, and accounts for substitutions, deletions and insertions, as well as rate heterogeneity among lineages as well as sites. However, its state space is limited to nucleotides, amino acids and codons; it is unable to, for example, simulate coevolving states (Fletcher and Yang, 2009).
Here, we present the COMPletely Arbitrary Sequence Simulator (COMPASS), a tool for simulating evolution over any arbitrary discrete state space, under any time-reversible evolutionary model along a phylogenetic tree. COMPASS allows for simulation under established rate matrices, and allows for straightforward implementation of novel models of nucleotide, protein, or codon evolution, processes of coevolution between state pairs, or the evolution of phenotypic characters.
2 Materials and methods
COMPASS is implemented in Python 2.7, and is implemented as command-line program, with all modules used available as part of the source code. It utilizes the Phylo module of BioPython (Talevich et al., 2012) to traverse trees, and a jump chain to simulate evolution of characters on a site-by-site basis (Yang, 2006). User-provided matrices are normalized so that branch lengths are equal to the expected number of substitutions. The rate matrices used by COMPASS can contain absolutely any symbols composed of ASCII characters (i.e. the user is not limited to nucleotides/proteins/codons), and the transition parameters can be any combination of frequencies and other completely arbitrary multipliers.
Standard options such as continuous and discrete gamma distribution of rates, invariant sites, partitioning of data into different models across both sites and branches, and rate heterogeneity among branches are available. Additionally, COMPASS can accept many standard tree formats (newick, nexus and phyloxml) for input, as well as outputting in a number of standard sequence alignment formats (clustal, fasta, phylip, phylip-sequential and stockholm, as well as tab-separated). This allows for many rich models of evolution to be implemented, such as mutation-selection models with either constant population size or varying population sizes over different branches of the tree. Full details are given in the user manual, which can be found with the Supplementary Information, or at labs.carleton.ca/eme/software-and-data.
3 Verification
COMPASS has been validated to ensure that the parameters entered will be accurately reflected in the simulated alignments. All parameters are reliably recovered. Data for nucleotide exchangeabilities, equilibrium frequencies, total branch length of input trees and dN/dS are shown in Figure 1. The dN/dS ratio was recovered using the codeml module of PAML 4.8 (Yang, 2007) under an M0 model, while actual values for branch lengths, exchangeabilities and equilibrium were recovered by running PhyloBayes MPI (Lartillot et al., 2013) for 1500 generations, taking the first 300 iterations as burn-in. Full details can be found in the Supplementary Information.
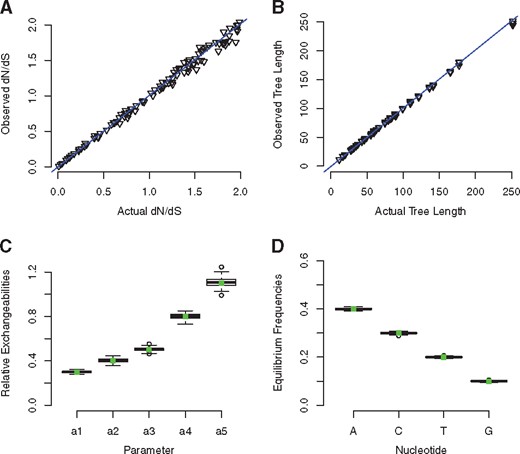
(A) Actual versus observed values of the dN/dS ratio. As expected, at lower (more realistic) values, correlation is extremely tight, while the relationship is noisier at higher values of dN/dS. Tests were carried out with a GY94-style matrix, a 50 species tree and 150 codons. (B) Actual versus observed total branch length of trees input into COMPASS. COMPASS very accurately simulates the total tree length. Sequences were evolved under a GTR model with 1000 sites. (C) Observed relative exchangeabilities for 130 sequences with 1000 sites under a GTR model (the sixth exchangeability was set to 1 and used to benchmark the others). Squares in centre of boxes represent actual values. 100 replicates were carried out. (D) Observed equilibrium frequencies for 130 sequences with 1000 sites, evolved under a GTR model. Squares in centre of boxes represent actual values. 100 replicates were carried out
4 Example usage: coevolution simulation
One area for which COMPASS is well-suited is in creating coevolution data. Identification of coevolving residues can contribute to protein structure prediction, RNA secondary structure prediction, identification of interacting sites between proteins, and protein interaction and protein complex predictions (Shang et al., 2012; Taylor et al., 2012; Weigt et al., 2009). However, there are few tools available to simulate coevolution data. PHASE (Gowri-Shankar and Rattray, 2007) can simulate coevolution for nucleotides, but not for amino acids or codons. MSAvolve (Ackerman et al., 2012) simulates coevolving protein data, but this evolution is not simulated along a tree, rendering it less useful for testing phylogenetically informed methods. COMPASS addresses all of these gaps.
The first step to using COMPASS is the creation of a parameter file that contains the frequencies of whatever character states one wishes to use, as well as a rate matrix specifying exchangeabilities between character states. These parameter files are simple text files that can be created in any text editor, allowing anybody to create completely new models or any reimplementation of existing models. In the case of nucleotide coevolution, a 16 by 16 rate matrix would be created, while for amino acid and codon coevolution 400 by 400 and 3721 by 3721 matrices, respectively, would be created. Complete descriptions and examples of these files and how to create them can be found in the Supplementary Information. Once parameter files have been created, COMPASS can simulate with the same model across all sites and branches, different models across sites, different models across branches, or any combination of models across sites and branches.
As COMPASS is implemented in Python, it is a tool that the bioinformatics community will easily be able to customize and extend as needed. As COMPASS simulates evolution site-by-site it would, for example, be relatively simple to parallelize it in order to enable fast simulation of very long (i.e. genome length) sequences, and use in Approximate Bayesian Computation (ABC) or Posterior Predictive Simulations (PPS). In addition, functionality for the creation of insertions and deletions could be added, making COMPASS even more usable in general cases as well as situations in which custom rate matrices are required. COMPASS fills a gap in current sequence simulation methods by being fully customizable and easy to use, giving users the power to quickly create and test new models of evolution, as well as simulating coevolution and any other process that can be modelled using discrete characters.
Acknowledgements
The authors would like to thank Tom Kazmirchuk for proofreading help, and Sebastian Hohna and an anonyomous reviewer for many helpful comments and suggestions.
Funding
This work was supported by an NSERC Discovery Grant (435248) and a CIHR New Investigator Salary Award (141995) to A.W., and an NSERC Discovery Grant (315924) to N.R.
Conflict of Interest: none declared.
References


{kind=link}