





Abstract
Motivation: Cysteine-rich proteins cover many important families in nature but there are currently no methods specifically designed for modeling the structure of these proteins. The accuracy of disulfide connectivity pattern prediction, particularly for the proteins of higher-order connections, e.g. >3 bonds, is too low to effectively assist structure assembly simulations.
Results: We propose a new hierarchical order reduction protocol called Cyscon for disulfide-bonding prediction. The most confident disulfide bonds are first identified and bonding prediction is then focused on the remaining cysteine residues based on SVR training. Compared with purely machine learning-based approaches, Cyscon improved the average accuracy of connectivity pattern prediction by 21.9%. For proteins with more than 5 disulfide bonds, Cyscon improved the accuracy by 585% on the benchmark set of PDBCYS. When applied to 158 non-redundant cysteine-rich proteins, Cyscon predictions helped increase (or decrease) the TM-score (or RMSD) of the ab initio QUARK modeling by 12.1% (or 14.4%). This result demonstrates a new avenue to improve the ab initio structure modeling for cysteine-rich proteins.
Availability and implementation: http://www.csbio.sjtu.edu.cn/bioinf/Cyscon/
Contact: [email protected] or [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The methods of protein structure prediction can generally be grouped into template-based and template-free (or ab initio) modeling (Zhang, 2008). Since the topology of the protein structure can be decided by the contact maps, many efforts have been dedicated to predicting residue-residue contacts. In the template-based modeling where homologous templates can be detected from the PDB, residue contacts can be reliably derived from the template structures (Misura et al., 2006; Zhang and Skolnick, 2004a). But for the ab initio modeling targets that do not have homologous templates, the contact information must be predicted from sequence either by feature-based training (Cheng and Baldi, 2007; Wu and Zhang, 2008) or correlated mutations (Göbel et al., 1994; Jones et al., 2012; Sun et al., 2015). In the 11th CASP (Critical Assessment of protein Structure Prediction) experiment, the modeling of several New Fold targets reached an unprecedented resolution due to the successful integration of the ab initio contact predictions (Grishin, 2014). However, except for the targets that have a high number of homologous sequences which can be used for detecting the conserved mutations and contacts, the accuracy of ab initio contact predictions is in general too low to be useful for 3D structure determination (Wu et al., 2011). One feasible improvement is to decompose the contact map into different types of contacts that can be predicted more reliably, such as disulfide bonds.
Disulfide bonds are formed between cysteine residues, which are the only coded amino acids that have a reactive sulfhydryl group. This type of residue interaction can be formed between residues from either the same or different polypeptide chains, which usually induces long-range contacts along the protein sequence (Gromiha and Selvaraj, 1997). It has been widely acknowledged that the long-range contacts are crucial to protein structure modeling because they constrain the possible conformations and reduce the entropy of unfolded states (Harrison and Sternberg, 1994; Wu and Zhang, 2008). Thus, accurate prediction of the disulfide-bonding network should improve ab initio protein structure prediction (Chuang et al., 2003; Gupta et al., 2004).
Disulfide bonds are important for protein function and structural stability. For instance, the formation of disulfide bonds is a key posttranslational modification in numerous proteins (Winther and Thorpe, 2014). In dual oxidase proteins, disulfide bonds contribute importantly to protein–protein interactions (Meitzler et al., 2013). Previous studies have revealed that incorrectly formed disulfide bonds can be deleterious to both the function and stability of some proteins (Cloos and Christgau, 2002; Kénesi et al., 2003).
Due to the importance of disulfide bonds in both protein structural and functional studies, many computational methods have been developed for predicting their connectivity patterns from protein sequence, with the aim to identify the correct bonding of oxidized cysteine residues. This problem was first addressed via maximum weight perfect matching (Fariselli and Casadio, 2001), where the weight of each edge is equivalent to the contact potential between the two cysteine residues, which was derived by Monte-Carlo simulated annealing. Following this work, considerable efforts were devoted to machine learning-based ab initio approaches to predict the contact potential for improving the performance. Progress in this regard focuses on two directions:
Developing more powerful prediction algorithms: e.g. neural network (NN) (Fariselli et al., 2002), support vector machine (SVM) (Chen et al., 2006), kernel method (Vincent et al., 2008), correlated mutation analysis (Raimondi et al., 2015; Rubinstein and Fiser, 2008) and support vector regression (SVR) (Savojardo et al., 2011, 2013; Song et al., 2007);
Introducing novel feature representations: besides traditional global and local sequence-derived features, recent studies have shown that some features such as protein subcellular localization (Savojardo et al., 2011), correlated mutations (Savojardo et al., 2013) and context-based features (Yaseen and Li, 2013) can also improve the performance. In addition, feature selection methods such as Fisher score (Zhu et al., 2010) were proposed to overcome the high-dimensional problems and improve the prediction.
Different from the above ab inito approaches which perform predictions by only using the amino acid sequence information, the other trend is using the homology modeling techniques, where some prediction features are extracted from the modeled structures. For instance, the spatial distance between the cysteine residues in the modeled structure can be used as an encoding feature (Yu et al., 2015). Other studies in this trend include: Lin and Tseng (2010) and O'Connor and Yeates (2004). Despite this type of methods is promising considering the rapid increase of the deposited 3D structures in PDB, which may increase the possibility of finding good templates, they may still fail for large portion of proteins, which cannot find good templates in current PDB.
This study aims to further enhancing the disulfide bonds prediction by using only the amino acid sequence information. Although existing methods can predict disulfide connectivity patterns with reasonable accuracy, there are still challenging problems that seriously limit the prediction performance. One of the biggest challenges is the high order problem: sequences with more disulfide bonds are much more difficult to predict accurately than those with fewer bonds. In fact, the entire pattern prediction accuracy will be very low for sequences with more than five bonds. The reason is that a disulfide connectivity pattern represents a unique correct combination of all the disulfide bonds in the sequence, and it is correct only if all the independent bonds are correctly predicted. For a sequence with 5 bonds, there will be 945 different bond combinations (c.f. Equation 4), where only 1 arrangement is correct and the random prediction success rate is ∼0.1%. The situation will be even worse for many cysteine-rich proteins, which often contain more than five disulfide bonds.
To this end, we propose a new algorithmic improvement based on the idea of order reduction by first finding the most confident disulfide bonds. For example, if we can determine 1 confident bond from a query sequence of high order (e.g. 5 bonds), then the problem is reduced to finding the correct combination among 4 remaining bonds, which has only 105 candidate patterns, much less than the original 945. It is fulfilled by an effective confident bond detector based on the observation that some disulfide bonds are found aligned at the same positions in the multiple sequence alignments (MSA). The detected confident bonds will be eliminated from the following decision process to reduce the complexity. Then, a maximum weight graph matching approach will be used to determine the remaining bonds, where the weights are predicted using a statistical machine learning predictor. Our hierarchical system for predicting disulfide connectivity patterns is called Cysteine contact (Cyscon). The new order reduction characteristic enables Cyscon to achieve higher prediction accuracy than traditional approaches.
2 Materials and methods
2.1 Datasets
For a fair comparison of the performance, we employed the two benchmark datasets that have been used in previous studies, i.e. SPX dataset (Cheng et al., 2006) and PDBCYS dataset (Savojardo et al., 2011). The first dataset is used to evaluate our proposed method, and the second is used for comparing with other existing methods. In these two datasets, only intra-chain disulfide bonds are presented and the disulfide bond information is derived from SSBOND records in the PDB files. Each dataset is described in detail below.
2.1.1 SPX dataset
This dataset was taken from DIpro (Cheng et al., 2006), which includes 1018 protein sequences that were collected from the PDB. These sequences have at least 1 intra-chain disulfide bond. We evaluated our method on the SPX dataset using 10-fold cross-validation, where the 10-folds were downloaded from http://dislocate.biocomp.unibo.it/dislocate/. We only selected chains containing at least 2 and at most 5 disulfide bonds from the original dataset for model training, resulting in 428 protein sequences, where the pairwise sequence identity is below 25%. Besides, 36 chains with more than 5 disulfide bonds were selected for an independent test of high order proteins.
2.1.2 PDBCYS dataset
In order to compare with the recent state-of-the-art methods, we assessed our method on the PDBCYS dataset using the same 20-fold cross-validation as DISLOCATE (Savojardo et al., 2011). This dataset is homology reduced at the 25% sequence identity level, which contains 1797 protein sequences. In the PDBCYS dataset, 458 sequences have at least 1 intra-chain disulfide bond. Similar to previous studies, chains with at least 2 and at most 5 disulfide bonds were picked from the original dataset, which resulting in 263 protein sequences for cross-validation. Additionally, 51 chains with more than 5 disulfide bonds were selected for an extra independent test. Supplementary Table S1 shows the detailed information of the two datasets.
2.2 System architecture
Cyscon predicts the disulfide bonds directly from amino acid sequence and is a hierarchical two-stage approach that integrates the machine learning-based predictions with the predictions from the sequence alignment-based confident bond detector (Fig. 1). Given a protein sequence, before the prediction of disulfide bond locations, we need to know the bonding states of each cysteine residue. The reason is that although disulfide bonds are only formed between cysteine residues, not necessarily every cysteine will be involved. Due to predicting whether a cysteine residue will be bonded or not can be transformed into a two-class classification problem and its accuracy is already very high (Chen et al., 2004), we assume that the bonding states of cysteine residues are known as the prior knowledge, which is the same as other previous studies (Rubinstein and Fiser, 2008; Song et al., 2007). In Cyscon, the confident bond detector assigns the most reliable disulfide bonds to the test sequence in the first stage, which will be eliminated from the undirected weighted graph for final decision. In the second stage, the sequential features of the oxidized cysteine residues are extracted and combined, and then fed into an SVR model to obtain the initial probabilities of cysteine pairs forming disulfide bonds. The first step can effectively reduce the decision complexity and hence is helpful for improving the predictor’s performance.

A schematic diagram of Cyscon to predict disulfide connectivity patterns. (1) highly conserved confident bond detector; (2) contact potential predicted by statistical machine learning-based engine; and (3) order reduction-based decision system
2.3 Feature extraction
Feature representation is critical in machine learning-based applications. In this work, we extracted four types of discriminative features to encode cysteine residues, which are all extracted directly from the amino acid sequence. The features are described in detail as follows.
2.3.1 Position specific scoring matrix (PSSM)
2.3.2 Predicted secondary structure (PSS)
The predicted secondary structure was also used to encode each cysteine residue by using PSIPRED (Jones, 1999), which outputs the propensities for the three secondary structure states (helix, strand and coil). To consider neighboring structural information, we encoded the PSS feature with a 13 × 3 = 39-D vector.
2.3.3 Correlated mutation (CM)
2.3.4 Cysteine separation distance (CSD)
where i and j represent the positions of the two oxidized cysteine residues along the query sequence that potentially form a disulfide bond. As shown in (Tsai et al., 2005), scaling the sequence distance value by the logarithm function works better than other scaling methods in terms of the accuracy of the final prediction.
In summary, according to the PSSM and PSS features, when the local window size is 13, we can get a vector of 13 × (20 + 3) × 2 = 598 components for each cysteine pair. Together with the CM (2D) and CSD (1D) features, we can then obtain a 601-D vector to encode one cysteine pair. We applied ε-insensitive support vector regression (ε-SVR), a module from the SVMlight (Joachims, 2002) software package to build the machine learning model using the radial basis function kernel (RBF) with ε = 0.01. The parameters γ and C were optimized by combining a grid search with cross-validation.
2.4 Confident bond detector
Based on the findings that sequences with similar cysteine separation profiles may have similar disulfide connectivity patterns (Zhao et al., 2005) and disulfide bonds are often highly conserved (Perlman et al., 1995), we built an effective confident bond detector engine. Given a protein sequence, we first used HHblits (Remmert et al., 2012) to search against the bundled UniProt20 database with three iterations to generate MSA. And then, we used HHsearch (Söding, 2005) to search against a database of profile hidden Markov models (HMMs), where the database was annotated with the known disulfide connectivity patterns. Lastly, we aligned the query sequence against the searched best sequence. Labeled disulfide bonds from the best aligned sequence are then assigned to the query sequence if the corresponding aligned positions on the query sequence are both cysteines. Our results show that disulfide bonds detected through this alignment-annotation approach have a high success rate and hence can be regarded as confident edges and then eliminated from the next-step of the decision process. This will help to reduce the decision order. Figure 2 illustrates the process of the confident bond detector. In the case of k-fold cross-validation, the annotated database is composed by the other k-1 training folds.
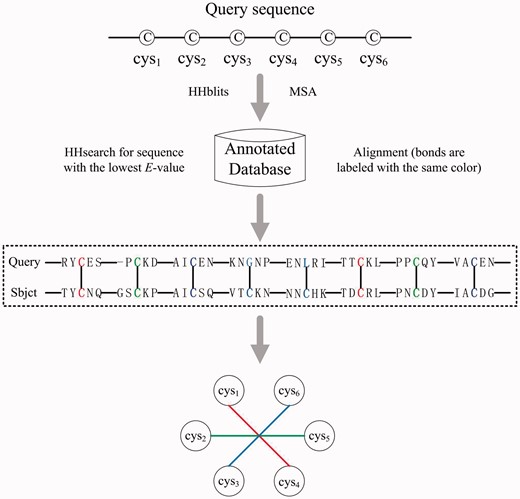
Flow chart of sequence alignment-based confident bond detector. By searching the query sequence against an annotated database of disulfide bonds, a confident bond is assigned between two cysteines when the following two conditions are satisfied: (1) their corresponding aligned residues are both cysteines, and (2) the two cysteines in the annotated sequence (Sbjct) form a disulfide bond. The detected confident bonds will be eliminated from the decision graph to reduce the problem order
2.5 Maximum weight graph matching
2.6 Application in QUARK-based ab initio structure prediction
QUARK is a recently developed method for ab initio protein structure prediction (Xu and Zhang, 2012). The structure fragments of 1–20 residues are first identified from unrelated protein structures by gapless matches. The full-length structure models are then constructed by reassembling the fragments using replica-exchange Monte Carlo simulations under the guidance of a composite physics- and knowledge-based force field, where the residue-based contacts derived from the distance profiles of short-range fragments are integrated as restraints to the force field (Xu and Zhang, 2013b). The QUARK method has been systematically tested in both blind CASP experiment (Xu et al., 2011) and genome-wide structure predictions (Xu and Zhang, 2013a) and demonstrated considerable advantage over peer methods on ab initio protein structure folding (Lee, 2012).
2.7 Evaluation criteria
The disulfide bond predictions are evaluated with two widely used criteria QC and QP, which are defined as QC = NC/TC and QP = NP/TP, respectively, where NC is the number of disulfide bonds that are correctly predicted, TC is the total number of disulfide bonds in the dataset, NP is the number of protein sequences whose disulfide connectivity patterns are correctly predicted and TP is the total number of protein sequences in the dataset.
The quality of the 3D structure models of the QUARK predictions is evaluated by the RMSD and TM-score (Zhang and Skolnick, 2004c), where the TM-score has been demonstrated to be more sensitive to the fold of protein structures, especially for very similar low-resolution structures that nevertheless still have high RMSD. A model with TM-score > 0.5 to native usually indicates correct modeling at the fold level based on large-scale statistics of PDB structures (Xu and Zhang, 2010).
3 Results
3.1 Evaluation of confident bond detector
In this work, we proposed a straightforward confident bond detector to reduce the decision order. This approach identifies disulfide bonds through searching the query sequence against a database with disulfide connectivity pattern annotations. We have tested this approach on three different annotated databases. The first is the benchmark SPX dataset, the second is benchmark PDBCYS dataset and the third is a bigger one called bDD collected from the recent release of Swiss-Prot database.
In the case of SPX dataset, we performed a 10-fold cross-validation, which uses HHsearch (Söding, 2005) to scan and match each of the test sequences against the annotated database that consists of the sequences from the other nine training folds. The best aligned sequence with the lowest E-value was selected to assign disulfide bonds to the query sequence using its annotation (Fig. 2). Detected disulfide bonds through this approach are very reliable by the fact that 301 from 322 assigned bonds are correct, which involves 173 tested protein sequences. The prediction accuracy of QC is as high as 93.5%. For the PDBCYS dataset, we performed 20-fold cross-validation. Finally, there are total 215 assigned bonds, and 199 bonds are correct, where 106 protein sequences are involved. The accuracy of QC is 92.6%.
We also evaluated this approach on a bigger annotated disulfide bonds database (bDD), which was constructed based on the newest Swiss-Prot database. This database was constructed according to the following procedures: (i) sequences with at least one disulfide bond have been collected; (ii) sequences annotated with ambiguous or uncertain terms, such as ‘potential’, ‘probable’, ‘probably’, ‘maybe’, ‘likely’ or ‘by similarity’ were excluded; (iii) inter-chain bonds were also excluded. Finally, we will obtain 3476 protein sequences in bDD. It’s important to note that in the testing process, we do not use any sequence in bDD that shares more than 25% sequence identity with the query sequence for strict evaluation purpose. By searching the SPX/PDBCYS dataset against bDD, the proposed confident bond detector assigned 455/285 bonds for 220/145 tested protein sequences on the two datasets, respectively, where 422/272 bonds are correct (QC = 92.7%/95.4%).
The above results demonstrate the following two interesting observations: (i) The coverage for bonds found by the confident bond detector can be improved when using a bigger annotated database. When tested on the SPX dataset with 10-fold cross-validation, the bond-based coverage is only 322/1265 = 25.5%, but this number increased to 455/1265 = 36.0% on the new bDD database. On the PDBCYS dataset, the coverage is increased from 215/804 = 26.7% to 285/804 = 35.4% when using bDD as the searching pool. (ii) Despite the different coverage ratios, all assigned disulfide bonds through the confident bond detector are very accurate. For instance, the QC will reach 92.6 and 95.4% on the PDBCYS and bDD databases, respectively. The results show that these detected bonds can be reliably removed from the undirected weighted graph to reduce the order of the model. Supplementary Tables S3–S5 show the performance at the different sequence identity thresholds.
3.2 Order reduction-driven hierarchical protocol enhances the prediction performance
Compared with the pure machine learning-based predictors, the new order reduction-driven hierarchical protocol is able to significantly improve the prediction accuracies. Tables 1 and 2 summarize the results obtained from the final consensus hierarchical protocol on the SPX and PDBCYS datasets, respectively. Taking the case of bDD as the searching pool as an example, on the SPX dataset, the Cyscon model can achieve the average prediction accuracies of QC and QP as 72.9 and 66.3%, respectively for B = 2–5 group, which are 10.9 and 9.0% higher than the pure machine learning-based SVR model. Similarly, on the PDBCYS dataset, QC and QP have been improved 10.8 and 13.0%.
Performance of disulfide connectivity pattern prediction on the SPX dataset
| numBa | SVRb | SVR + OR (SPX)c | SVR + OR (bDD)d | SVR + OR (bDD)d | SVR + OR (bDD)d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 71.4 | 71.4 | 75.8 | 75.8 | 73.3 | 73.3 | 78.0 | 78.0 | 77.0 | 77.0 |
| 3 | 61.8 | 70.3 | 64.8 | 73.3 | 62.0 | 70.8 | 63.1 | 72.1 | 67.8 | 75.6 |
| 4 | 44.3 | 57.0 | 56.7 | 66.7 | 45.5 | 58.0 | 54.3 | 66.7 | 55.4 | 70.2 |
| 5 | 11.5 | 45.2 | 36.4 | 62.4 | 19.8 | 52.1 | 23.6 | 54.1 | 36.3 | 61.9 |
| 2–5 | 57.3 | 62.0 | 65.4 | 71.2 | 60.5 | 65.9 | 64.1 | 69.7 | 66.3 | 72.9 |
| 6–10e | 11.1 | 32.8 | 11.1 | 34.8 | 11.1 | 34.0 | 16.7 | 40.4 | 22.2 | 43.6 |
| numBa | SVRb | SVR + OR (SPX)c | SVR + OR (bDD)d | SVR + OR (bDD)d | SVR + OR (bDD)d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 71.4 | 71.4 | 75.8 | 75.8 | 73.3 | 73.3 | 78.0 | 78.0 | 77.0 | 77.0 |
| 3 | 61.8 | 70.3 | 64.8 | 73.3 | 62.0 | 70.8 | 63.1 | 72.1 | 67.8 | 75.6 |
| 4 | 44.3 | 57.0 | 56.7 | 66.7 | 45.5 | 58.0 | 54.3 | 66.7 | 55.4 | 70.2 |
| 5 | 11.5 | 45.2 | 36.4 | 62.4 | 19.8 | 52.1 | 23.6 | 54.1 | 36.3 | 61.9 |
| 2–5 | 57.3 | 62.0 | 65.4 | 71.2 | 60.5 | 65.9 | 64.1 | 69.7 | 66.3 | 72.9 |
| 6–10e | 11.1 | 32.8 | 11.1 | 34.8 | 11.1 | 34.0 | 16.7 | 40.4 | 22.2 | 43.6 |
aNumber of bonds.
bPure SVR model trained with four types of sequential features.
cOrder reduction (OR)-driven prediction protocol. Confident bond detections were derived from 10-fold cross-validation on the SPX dataset.
dOrder reduction (OR)-driven prediction protocol. The bDD database was used and the searched sequence shares no more than the certain sequence identities with the query sequence (15, 20 and 25%, respectively).
eAverage results for proteins with the number of disulfide bonds from 6 to 10.
Performance of disulfide connectivity pattern prediction on the SPX dataset
| numBa | SVRb | SVR + OR (SPX)c | SVR + OR (bDD)d | SVR + OR (bDD)d | SVR + OR (bDD)d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 71.4 | 71.4 | 75.8 | 75.8 | 73.3 | 73.3 | 78.0 | 78.0 | 77.0 | 77.0 |
| 3 | 61.8 | 70.3 | 64.8 | 73.3 | 62.0 | 70.8 | 63.1 | 72.1 | 67.8 | 75.6 |
| 4 | 44.3 | 57.0 | 56.7 | 66.7 | 45.5 | 58.0 | 54.3 | 66.7 | 55.4 | 70.2 |
| 5 | 11.5 | 45.2 | 36.4 | 62.4 | 19.8 | 52.1 | 23.6 | 54.1 | 36.3 | 61.9 |
| 2–5 | 57.3 | 62.0 | 65.4 | 71.2 | 60.5 | 65.9 | 64.1 | 69.7 | 66.3 | 72.9 |
| 6–10e | 11.1 | 32.8 | 11.1 | 34.8 | 11.1 | 34.0 | 16.7 | 40.4 | 22.2 | 43.6 |
| numBa | SVRb | SVR + OR (SPX)c | SVR + OR (bDD)d | SVR + OR (bDD)d | SVR + OR (bDD)d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 71.4 | 71.4 | 75.8 | 75.8 | 73.3 | 73.3 | 78.0 | 78.0 | 77.0 | 77.0 |
| 3 | 61.8 | 70.3 | 64.8 | 73.3 | 62.0 | 70.8 | 63.1 | 72.1 | 67.8 | 75.6 |
| 4 | 44.3 | 57.0 | 56.7 | 66.7 | 45.5 | 58.0 | 54.3 | 66.7 | 55.4 | 70.2 |
| 5 | 11.5 | 45.2 | 36.4 | 62.4 | 19.8 | 52.1 | 23.6 | 54.1 | 36.3 | 61.9 |
| 2–5 | 57.3 | 62.0 | 65.4 | 71.2 | 60.5 | 65.9 | 64.1 | 69.7 | 66.3 | 72.9 |
| 6–10e | 11.1 | 32.8 | 11.1 | 34.8 | 11.1 | 34.0 | 16.7 | 40.4 | 22.2 | 43.6 |
aNumber of bonds.
bPure SVR model trained with four types of sequential features.
cOrder reduction (OR)-driven prediction protocol. Confident bond detections were derived from 10-fold cross-validation on the SPX dataset.
dOrder reduction (OR)-driven prediction protocol. The bDD database was used and the searched sequence shares no more than the certain sequence identities with the query sequence (15, 20 and 25%, respectively).
eAverage results for proteins with the number of disulfide bonds from 6 to 10.
Performance comparison with other methods on the PDBCYS dataset
| numBa | DISLOCATE | DMCb | SVRc | Cyscond | Cyscone | Cyscone | Cyscone | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 75 | 75 | 76.0 | 76.0 | 79.9 | 79.9 | 83.4 | 83.4 | 83.1 | 83.1 | 83.5 | 83.5 | 84.4 | 84.4 |
| 3 | 48 | 60 | 55.3 | 62.8 | 53.6 | 64.3 | 61.6 | 69.3 | 60.4 | 69.3 | 68.5 | 75.8 | 76.5 | 82.5 |
| 4 | 44 | 57 | 51.2 | 67.7 | 52.8 | 66.4 | 55.6 | 68.4 | 52.8 | 66.4 | 54.2 | 67.0 | 57.4 | 69.6 |
| 5 | 19 | 46 | 32.4 | 58.9 | 29.4 | 50.7 | 39.2 | 59.0 | 34.3 | 53.6 | 43.1 | 62.3 | 55.4 | 69.9 |
| 2–5 | 54 | 60 | 59.3 | 66.2 | 59.5 | 65.8 | 65.3 | 70.4 | 63.1 | 68.6 | 67.2 | 72.7 | 72.3 | 77.0 |
| 6–10f | – | – | – | – | 2.0 | 34.3 | 7.8 | 39.8 | 3.9 | 36.7 | 9.8 | 43.0 | 13.7 | 46.2 |
| numBa | DISLOCATE | DMCb | SVRc | Cyscond | Cyscone | Cyscone | Cyscone | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 75 | 75 | 76.0 | 76.0 | 79.9 | 79.9 | 83.4 | 83.4 | 83.1 | 83.1 | 83.5 | 83.5 | 84.4 | 84.4 |
| 3 | 48 | 60 | 55.3 | 62.8 | 53.6 | 64.3 | 61.6 | 69.3 | 60.4 | 69.3 | 68.5 | 75.8 | 76.5 | 82.5 |
| 4 | 44 | 57 | 51.2 | 67.7 | 52.8 | 66.4 | 55.6 | 68.4 | 52.8 | 66.4 | 54.2 | 67.0 | 57.4 | 69.6 |
| 5 | 19 | 46 | 32.4 | 58.9 | 29.4 | 50.7 | 39.2 | 59.0 | 34.3 | 53.6 | 43.1 | 62.3 | 55.4 | 69.9 |
| 2–5 | 54 | 60 | 59.3 | 66.2 | 59.5 | 65.8 | 65.3 | 70.4 | 63.1 | 68.6 | 67.2 | 72.7 | 72.3 | 77.0 |
| 6–10f | – | – | – | – | 2.0 | 34.3 | 7.8 | 39.8 | 3.9 | 36.7 | 9.8 | 43.0 | 13.7 | 46.2 |
aNumber of bonds.
bDISLOCATE + MIp + iCOV.
cPure SVR model trained with four types of sequential features.
dConfident bond detections were derived from 20-fold cross-validation on the PDBCYS dataset.
ebDD was used as the searching pool and the searched sequence shares no more than the certain sequence identities with the query sequence (15, 20 and 25%, respectively).
fOverall results for proteins with the number of disulfide bonds from 6 to 10, where no results reported in DISLOCATE and DMC for this group.
Performance comparison with other methods on the PDBCYS dataset
| numBa | DISLOCATE | DMCb | SVRc | Cyscond | Cyscone | Cyscone | Cyscone | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 75 | 75 | 76.0 | 76.0 | 79.9 | 79.9 | 83.4 | 83.4 | 83.1 | 83.1 | 83.5 | 83.5 | 84.4 | 84.4 |
| 3 | 48 | 60 | 55.3 | 62.8 | 53.6 | 64.3 | 61.6 | 69.3 | 60.4 | 69.3 | 68.5 | 75.8 | 76.5 | 82.5 |
| 4 | 44 | 57 | 51.2 | 67.7 | 52.8 | 66.4 | 55.6 | 68.4 | 52.8 | 66.4 | 54.2 | 67.0 | 57.4 | 69.6 |
| 5 | 19 | 46 | 32.4 | 58.9 | 29.4 | 50.7 | 39.2 | 59.0 | 34.3 | 53.6 | 43.1 | 62.3 | 55.4 | 69.9 |
| 2–5 | 54 | 60 | 59.3 | 66.2 | 59.5 | 65.8 | 65.3 | 70.4 | 63.1 | 68.6 | 67.2 | 72.7 | 72.3 | 77.0 |
| 6–10f | – | – | – | – | 2.0 | 34.3 | 7.8 | 39.8 | 3.9 | 36.7 | 9.8 | 43.0 | 13.7 | 46.2 |
| numBa | DISLOCATE | DMCb | SVRc | Cyscond | Cyscone | Cyscone | Cyscone | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | QP | QC | |
| 2 | 75 | 75 | 76.0 | 76.0 | 79.9 | 79.9 | 83.4 | 83.4 | 83.1 | 83.1 | 83.5 | 83.5 | 84.4 | 84.4 |
| 3 | 48 | 60 | 55.3 | 62.8 | 53.6 | 64.3 | 61.6 | 69.3 | 60.4 | 69.3 | 68.5 | 75.8 | 76.5 | 82.5 |
| 4 | 44 | 57 | 51.2 | 67.7 | 52.8 | 66.4 | 55.6 | 68.4 | 52.8 | 66.4 | 54.2 | 67.0 | 57.4 | 69.6 |
| 5 | 19 | 46 | 32.4 | 58.9 | 29.4 | 50.7 | 39.2 | 59.0 | 34.3 | 53.6 | 43.1 | 62.3 | 55.4 | 69.9 |
| 2–5 | 54 | 60 | 59.3 | 66.2 | 59.5 | 65.8 | 65.3 | 70.4 | 63.1 | 68.6 | 67.2 | 72.7 | 72.3 | 77.0 |
| 6–10f | – | – | – | – | 2.0 | 34.3 | 7.8 | 39.8 | 3.9 | 36.7 | 9.8 | 43.0 | 13.7 | 46.2 |
aNumber of bonds.
bDISLOCATE + MIp + iCOV.
cPure SVR model trained with four types of sequential features.
dConfident bond detections were derived from 20-fold cross-validation on the PDBCYS dataset.
ebDD was used as the searching pool and the searched sequence shares no more than the certain sequence identities with the query sequence (15, 20 and 25%, respectively).
fOverall results for proteins with the number of disulfide bonds from 6 to 10, where no results reported in DISLOCATE and DMC for this group.
It can be also observed from Tables 1 and 2 that when we used the bigger annotated database bDD as the searching pool, the results are all better than the cases of cross-validation searching the smaller SPX and PDBCYS datasets. The reason is that the coverage of detected confident bonds is increased as well when using the bigger bDD. It’s interesting to observe that the improvement is higher on the PDBCYS dataset. For instance, QP is improved from 65.4 to 66.3% (0.9%) for B = 2–5 group on the SPX dataset, while on the PDBCYS dataset, QP is improved from 65.3 to 72.3% (7.0%). To dig the reason, we calculated the overlap ratios between the confident bonds searched from different annotated pools and those bonds with the largest probability predicted by the SVR model. On the SPX dataset, the overlap ratios are 23.6 and 23.5% when using the smaller SPX and bigger bDD, respectively, which are very close. On the contrary, for the PDBCYS dataset, the two ratios are 31.6 and 27.7%, respectively. In the maximum weight graph matching step, the predicted bond with the largest probability may be selected as a component bond of connectivity pattern in a greedy way. Thus, the lower the overlap ratio is, the more helpful of the detected confident bonds to the final system performance due to the more diversity generated in the consensus system. Hence, on the PDBCYS dataset, using bDD is more helpful since the overlap ratio is 3.9% lower than the smaller pool, whereas this number is 0.1% on the SPX dataset.
One of the most challenging problems in disulfide connectivity pattern prediction is the high order problem. For example, if there are 6 bonds in the protein sequence, we will have to find the unique real connectivity pattern from 10 395 candidates (Equation 4), and the random assignment correctness ratio will be as low as 0.096‰. This extreme imbalance significantly limits the feasibility of the maximum weight perfect matching approach. Motivated by the idea of reducing the problem order, we eliminate the detected confident bonds in the first layer. Through this way, the possible solutions can be decreased exponentially and the problem can be thus solved much more easily. From Tables 1 and 2, we can already see that the performance is improved as expected for the group of B = 2–5.
In order to demonstrate our approach for dealing with higher order proteins, we collected 36 and 51 proteins with B = 6–10 bonds from the SPX and PDBCYS datasets for independent tests, respectively. Tables 1 and 2 show Cyscon’s results on the higher order group of B = 6–10. As shown in Table 1, when we used the pure SVR model on SPX, only 4 out of 36 proteins’ connectivity patterns can be correctly predicted (QP = 11.1%). While the number was increased to eight if we introduced the order reduction idea by using bDD as the searching pool (QP = 22.2%). Our results demonstrate that the order-reduction approach is especially useful for reducing the order. For instance, the test protein of 1olzA has eight disulfide bonds. The traditional pure SVR prediction model has to determine the unique pattern from 2 027 025 candidates, which failed. However in the Cyscon protocol, 5 confident bonds have been identified in the first layer, which has significantly reduced the original searching problem to much less 15 candidates, which is finally correctly predicted.
Similarly, for the 51 proteins of B = 6–10 in the PDBCYS dataset, only 1 connectivity pattern can be correctly predicted when using the pure SVR model (QP = 2.0%). This serious situation was relieved by using the order reduction. Concretely, 6 more proteins can be correctly predicted (QP = 13.7%), i.e. 3 proteins with 6 bonds, 1 protein with 7 bonds and 2 proteins with 8 bonds. These results demonstrate the effectiveness of the order reduction strategy in Cyscon.
3.3 Comparison with existing predictors
To compare with the recent state-of-the-art methods (Savojardo et al., 2011, 2013), we also evaluated our method on the benchmark PDBCYS dataset using the same 20-fold cross-validation and also the same folds as previous studies. This dataset is homology reduced at the 25% sequence identity level. Table 2 shows the results. As can be seen, the Cyscon model gives better results than DISLOCATE (Savojardo et al., 2011), which was trained with the additional feature of subcellular localization. It also outperforms DMC (DISLOCATE + MIp + iCOV) (Savojardo et al., 2013), which was trained with additional correlated mutation features calculated by using MIp (Dunn et al., 2008) and PSICOV (Jones et al., 2012). The overall accuracies of QC and QP of final Cyscon are 77.0 and 72.3%, respectively, which are 10.8 and 13.0% higher than DMC. The power of the newly developed Cyscon on the high order proteins is obvious. Taking the subset of B = 5 for instance (compared to B = 4), QP drops 25% accuracy in DISLOCATE and 18.8% in DMC, while Cyscon drops only 2.0%. This also demonstrates the efficacy of the proposed order reduction approach for high order problems. It’s also worthy to mention that Cyscon detected 285 confident bonds in the first layer on the PDBCYS dataset, and these bonds will not be participated in the final maximum weight matching decision process, which is one of the major reasons for the performance improvement.
3.4 Case study
To highlight why Cyscon works better, we take the protein 1jxcA as an example. There are 4 native disulfide bonds in this protein, i.e. C11–C63, C24–C48, C33–C58 and C37–C60. Hence, there will be 105 possible combination patterns in this sequence. For the pure machine learning-based SVR model, it mistakenly predicted the disulfide connectivity pattern as: C11–C33, C24–C48, C37–C58 and C60–C63 (Fig. 3). Thus, only the second bond is correct, which makes QC = 12.5% and QP = 0% on this protein.

Illustration of Cyscon on the case protein of 1jxcA. Bold lines indicate the observed or predicted disulfide bonds. SVR tries to detect the pattern in all 105 possible combination candidates. In Cyscon, by removing 2 confident edges, it tries to detect the pattern in 3 possible combination candidates
However, in the final confident bond-driven Cyscon predictor, the confident bond detector found 2 confident bonds as C33–C58 and C37–C60. We thus eliminated the two confident edges from the undirected weighted graph. Then we performed maximum perfect matching algorithm on the order-reduced weighted graph composed of only four cysteine residues, which has only three possible combination patterns. Expectedly, we obtained a correct sub connectivity pattern of C11–C63 and C24–C48. Together with the two steps, Cyscon predicted the correct connectivity pattern QC = 100% and QP = 100% for the protein 1jxcA as illustrated in Figure 3.
3.5 Application to 3D structure modeling of cysteine-rich proteins
There are many cysteine-rich proteins, such as from various toxins etc. A common feature of the cysteine-rich proteins is that there are many disulfide bonds. Although the structure prediction field has achieved great successes in the past decade, there are few structural modelers that are specifically developed for the cysteine-rich proteins. A typical feature for the cysteine-rich protein 3D structure predictors is that they should be capable of incorporating the disulfide bonds as restraints. We have obtained statistics on 105 339 entries in the current PDB database, where 15 429 structures are found to have at least 3 disulfide bonds (∼14.6%). The high ratio also indicates the importance of adding the disulfide connectivity pattern into the structure prediction, especially considering that most of them are long-range contacts.
To examine quantitatively the usefulness of the predicted disulfide connectivity patterns to protein 3D structure prediction, we collected 158 protein sequences (see supplementary Table S2) from the PDB with experimentally determined 3D structures according to the following criteria: (i) solved by X-ray diffraction; (ii) resolution less than 2.5 Å; (iii) number of bonds not less than 3; (iv) number of residues not less than 20 and not more than 100; and (v) sequence identity not more than 30%. We focused on the small proteins below 100 residues because the number of the disulfide bond pairs is relatively low (3–5), compared with the coverage of contacts normally needed to derive the tertiary structure (>L/10) (Li et al., 2004; Wu and Zhang, 2008; Zhang et al., 2003).
We first predicted the disulfide connectivity patterns for the 158 proteins from their primary sequence with the above Cyscon predictor; then, we used the ab initio modeling software QUARK (Xu and Zhang, 2012) to predict their 3D structures in two cases: (i) without restraints from the connectivity patterns; and (ii) with restraints from the predicted connectivity patterns. The average results are listed in Table 3.
Structure modeling of 158 proteins by QUARK with or without disulfide bonds predicted by Cyscon as restraints
| numBa | 3 | 4 | 5 | >5 | Overall |
|---|---|---|---|---|---|
| numPb | 83 | 37 | 23 | 15 | 158 |
| TM-scoreQUAc | 0.303 | 0.279 | 0.255 | 0.212 | 0.282 |
| TM-scoreCysc | 0.343 | 0.300 | 0.293 | 0.240 | 0.316 |
| RMSDQUAd | 8.9 | 9.7 | 11.2 | 11.3 | 9.7 |
| RMSDCysd | 7.2 | 8.7 | 9.9 | 10.4 | 8.3 |
| numBa | 3 | 4 | 5 | >5 | Overall |
|---|---|---|---|---|---|
| numPb | 83 | 37 | 23 | 15 | 158 |
| TM-scoreQUAc | 0.303 | 0.279 | 0.255 | 0.212 | 0.282 |
| TM-scoreCysc | 0.343 | 0.300 | 0.293 | 0.240 | 0.316 |
| RMSDQUAd | 8.9 | 9.7 | 11.2 | 11.3 | 9.7 |
| RMSDCysd | 7.2 | 8.7 | 9.9 | 10.4 | 8.3 |
aNumber of bonds.
bNumber of proteins.
cTM-score of QUARK prediction without Cyscon predictions (TM-scoreQUA), and with Cyscon predictions (TM-scoreCys).
dRMSD (Å) of QUARK prediction without Cyscon predictions (RMSDQUA), and with Cyscon predictions (RMSDCys).
Structure modeling of 158 proteins by QUARK with or without disulfide bonds predicted by Cyscon as restraints
| numBa | 3 | 4 | 5 | >5 | Overall |
|---|---|---|---|---|---|
| numPb | 83 | 37 | 23 | 15 | 158 |
| TM-scoreQUAc | 0.303 | 0.279 | 0.255 | 0.212 | 0.282 |
| TM-scoreCysc | 0.343 | 0.300 | 0.293 | 0.240 | 0.316 |
| RMSDQUAd | 8.9 | 9.7 | 11.2 | 11.3 | 9.7 |
| RMSDCysd | 7.2 | 8.7 | 9.9 | 10.4 | 8.3 |
| numBa | 3 | 4 | 5 | >5 | Overall |
|---|---|---|---|---|---|
| numPb | 83 | 37 | 23 | 15 | 158 |
| TM-scoreQUAc | 0.303 | 0.279 | 0.255 | 0.212 | 0.282 |
| TM-scoreCysc | 0.343 | 0.300 | 0.293 | 0.240 | 0.316 |
| RMSDQUAd | 8.9 | 9.7 | 11.2 | 11.3 | 9.7 |
| RMSDCysd | 7.2 | 8.7 | 9.9 | 10.4 | 8.3 |
aNumber of bonds.
bNumber of proteins.
cTM-score of QUARK prediction without Cyscon predictions (TM-scoreQUA), and with Cyscon predictions (TM-scoreCys).
dRMSD (Å) of QUARK prediction without Cyscon predictions (RMSDQUA), and with Cyscon predictions (RMSDCys).
From Table 3, we can see that the connectivity pattern restraints improve the accuracy of protein 3D structure modeling. Despite the low coverage of the disulfide bond predictions relative to the normal contact predictions, the results show an average improvement of 12.1% for TM-score and 14.4% for RMSD by the introduction of the Cyscon predictions. There are 10 protein models that improved from TM-score below 0.5 to above 0.5 as shown in Table S2 (1edmB, 1hypA, 1n69A, 1p9gA, 2posA, 2rjiA, 3qteA, 3s64A, 3tvjI and 4i6oA).
In Figure 4, we show three examples of QUARK versus Cyscon-assisted QUARK modeling of 3qteA, 1p9gA and 4i6oA, respectively. First, 3qteA is an antimicrobial protein with 32 residues, where Cyscon generated 3 disulfide bond predictions which are all correct. The first model by the original QUARK simulation without Cyscon only has 1 disulfide bond satisfied which resulted in an incorrect fold with TM-score = 0.166. When integrated with the Cyscon predictions, 3 Cyscon contacts were satisfied and the TM-score of the first QUARK model was increased to 0.572 (Fig. 4a). The second example from 1p9gA is an antifungal protein with 40 residues and 5 disulfide bond predictions by Cyscon. Again, the inclusion of the Cyscon contacts increased the number of disulfide bond satisfaction from 2 to 4, and therefore the TM-score of the first model improved from 0.206 to 0.508 (Fig. 4b). The final example is an alpha protein from 4i6oA with 68 residues. Cyscon generated 3 disulfide bonds with an accuracy = 100%, where 0 of them are satisfied in the original QUARK model. The Cyscon-assisted QUARK model has 3 predicted disulfide bonds satisfied which brought the TM-score of the first model from 0.286 to 0.676 (Fig. 4c).
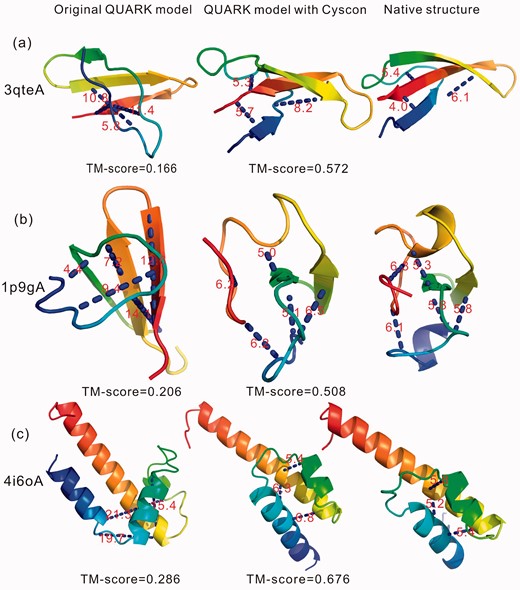
Illustrative examples of QUARK modeling improved by Cyscon disulfide bond predictions. From left to right columns: the first QUARK model without Cyscon, the first QUARK model with Cyscon predictions, and the experimental structures. The dot blue lines label the disulfide bonds predicted by Cyscon with number denoting the actual distance in the structures. (a) 3qteA; (b) 1p9gA; (c) 4i6oA
4 Discussion
One challenge to the ab initio prediction of disulfide connectivity pattern is the high order problem, i.e. the prediction accuracy will drop significantly with the increasing of number of disulfide bonds. The mathematical reason to the issue is that the number of possible disulfide connectivity patterns increases exponentially with the number of Cyscine residues (Supplementary Fig. S1). It consequently induces the high order of graph search in the maximum weight graph matching. Motivated by the straightforward idea of order reduction, we developed a novel disulfide bond predictor, Cyscon, which successfully reduces the graph matching order by eliminating the highly confident disulfide bonds, which are detected by a newly designed detector.
The high order problem can be effectively solved by the proposed approach of this paper, resulting in significant improvement of the prediction performance. For instance, on the SPX dataset, the residue-based accuracy (QC) and the protein-based accuracy (QP) in the subset of test proteins with 5 bonds are 16.7 and 24.8% higher, respectively when comparing the final Cyscon predictor to the pure machine learning-based SVR method. For the proteins with more than five bonds, QP is two times better than the original performance. On the PDBCYS dataset, QP is 6.9 times better than the pure SVR model for the proteins of 6–10 bonds.
Inspired by the improvements from sequence alignment-based order reduction, we have also tried to remove the edge with the highest weight corresponding to machine learning-based contact potential for comparison. However, the results are much worse. For the 428 protein sequences in the SPX dataset, we found that QC is 284/428 = 66.4%, which is far worse than the proposed confident edge detector. The reason is probably due to the noise from SVR predictions which has a lower confidence than the stringent sequence-alignment derivations.
Although the order reduction approach can improve the prediction performance, we found that the improvement depends on the detection coverage. Our experimental results show that when using the big bDD as the searching pool, the improvement is generally higher. We will keep on updating bDD database to include the most up-to-date data for improving the confident bond detection success rate, which is expected to further enhance the overall prediction accuracy for high order protein sequences.
Besides the high order problem, inter-chain disulfide bond prediction is another important issue need to be addressed. However, there are few studies devoted to the problem probably due to the fact that the annotated inter-chain bonds are very few in the current databases. This type of bond is related to protein quaternary structure and protein–protein interactions. Hence, predicting inter-chain disulfide bonds will be an important follow-up work, where similar idea of order reduction might expect to be helpful.
We also show that predicting reliable disulfide connectivity patterns can improve the 3D structure modeling of cysteine-rich proteins. Using QUARK (Xu and Zhang, 2012) as an example, our data show that the quality of ab initio structure predictions can be significantly improved when adding the predicted disulfide connectivity patterns as distance restraints. This is consistent with the exciting successes on contact-driven protein structure prediction recently observed in the CASP 11 experiment (Grishin, 2014) and other contact-assisted structure prediction efforts (Marks et al., 2011; Wu et al., 2011).
Funding
This work was supported in part by the National Natural Science Foundation of China (61222306, 91130033, 61175024), Shanghai Science and Technology Commission (11JC1404800), and the National Institute of General Medical Sciences (GM083107).
Conflict of Interest: none declared.
References
Author notes
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
Associate Editor: Anna Tramontano


{kind=link}
{kind=link}
{kind=link}
{kind=link}