





Abstract
Motivation: Comprehensive analysis of genome-wide molecular data challenges bioinformatics methodology in terms of intuitive visualization with single-sample resolution, biomarker selection, functional information mining and highly granular stratification of sample classes. oposSOM combines those functionalities making use of a comprehensive analysis and visualization strategy based on self-organizing maps (SOM) machine learning which we call ‘high-dimensional data portraying’. The method was successfully applied in a series of studies using mostly transcriptome data but also data of other OMICs realms.
Availability and implementation: oposSOM is now publicly available as Bioconductor R package.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Bioinformatics tools are needed which allow to statistically, functionally and visually summarise high-dimensional data such as transcriptome studies at different levels of resolution ranging from individual samples and genes to sample classes and expression modules of co-regulated genes. For this purpose, we developed a bioinformatics analysis pipeline based on self-organizing map (SOM) machine learning which facilitates a holistic view on this data (Wirth et al., 2011, 2012b). We termed this technique ‘high-dimensional data portraying’. It subsumes the visualization of the data landscape of each individual, a series of downstream bioinformatics and statistics analysis options and the detailed and comprehensive reporting of the results. We have chosen SOM machine learning as backbone because it combines strong clustering, dimension reduction, multidimensional scaling and visualization capabilities which have been shown to be advantageous compared to alternative methods such as clustering heatmaps and negative matrix factorization when applied to molecular high-throughput data (see Wirth et al., 2011 and references cited therein). We complemented the basal SOM algorithm with a sophisticated data analysis workflow including visualization of the individual feature landscapes, statistical testing for differential features and biomarker selection, mining of biological function, and also sample diversity analysis to assess classes of samples. oposSOM continues and largely extends the scope of a previous SOM-based expression analysis tool, the ‘gene expression dynamic inspector’ (GEDI) (Eichler et al., 2003): oposSOM is under steady development, provides a multitude of sample diversity analyses and, most importantly, provides comprehensive functional annotations.
Our portraying-method has been developed in first instance for gene expression data comprising from tens up to thousands of samples (e.g. tumour specimen in patient cohorts, experimental conditions in cell line experiments). The portraying functionality is unique and suited especially for scientists who attach importance to visual control and intuitive perception of complex data. The software was applied in a series of previous studies aiming at discovering the gene expression landscapes of healthy human tissues (Wirth et al., 2011), of cancer subtypes (Hopp et al., 2013a, b; Reifenberger et al., 2014) and of stem cell development (Charbord et al., 2014). Further applications addressed the integrative analysis of mRNA and miRNA expression data (Cakir et al., 2014), the proteome of algae (Wirth et al., 2012a), whole genome histone modification patterns (Steiner et al., 2012) and the genomic diversity of human ethnicities (Binder and Wirth, 2015).
2 Functionality
2.1 Package usability
The oposSOM package requires the input of gene-centered expression data solely, e.g. as pre-processed microarray intensity data or RNA-seq read counts in log-scale. All other program parameters are optional (see package vignette). An image of the analysis environment is stored upon completion of the oposSOM run.
2.2 Workflow
oposSOM comprises a multitude of analysis modules whose functionalities were described in detail in our previous publications. An illustration of the workflow and a complete list of methods implemented in the package can be found in the Supplementary Material. In brief, the package fulfils the following tasks:
The SOM space obtained from the training process is characterized by several supporting maps and profiles providing, e.g. the number of genes mapped to each meta-gene.
Samples are individually portrayed in PDF report sheets allowing the detailed examination of their expression landscapes and especially to identify modules of co-expressed genes.
Feature maps, reports and lists allow feature selection and evaluation of their statistical significance.
Gene set enrichment analysis of the expression modules provides their functional context based on a large collection of predefined gene sets.
Sample diversity analysis and class discovery is performed using multiple algorithms (e.g. hierarchical clustering, correlation spanning tree) and different metrics (Euclidean distance, Pearson’s correlation coefficient).
2.3 Results
oposSOM stores the results in a defined folder structure. These results comprise a variety of PDF documents, which provide extensive information about the systems studied (for example plots and images of the input data, supplementary descriptions of the SOM generated and associated metadata, the sample diversity landscape and also functional annotations). The PDF reports are complemented by CSV spreadsheets, which render the complete information accessible. Detailed descriptions of the algorithms and visualizations were given in our previous publications (Hopp et al., 2013a, b; Wirth, 2012; Wirth et al., 2011, 2012b). HTML files are generated to provide easy access to the analysis results via an intuitive and descriptive interface. A Summary.html can be found in the results folder created by oposSOM. We recommend new users to browse the results using this interface.
3 Use case: portraying of cancer subtypes
We applied oposSOM to patient expression data of mature aggressive B-cell lymphomas to characterize their genome wide expression landscapes in terms of four distinct molecular subtypes which associate with differing clinical phenotypes and survival prognosis (Hopp et al., 2013a).
Figure 1 provides an overview of the analysis steps: The expression portraits visualize the expression landscape of each individual sample (Fig. 1a) and of each subtype (Fig. 1b). Red and blue ‘spots’ in the portraits can be assigned to modules of co-expressed genes up- and down-regulated in the respective sample/subtype, respectively. The subtype portraits in Figure 1b immediately reveal distinct and subtype-specifically over-expressed expression modules emerging as red spots located near the corners of the respective portrait.
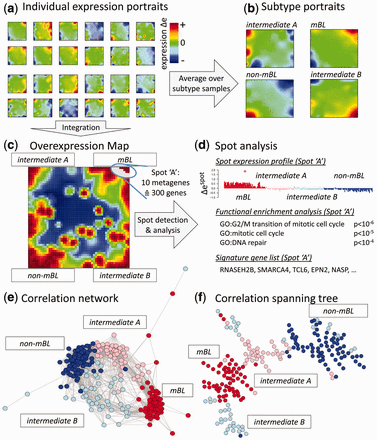
oposSOM analysis of a cohort of 220 mature B-cell lymphoma cases (see text)
All expression modules detected are summarized in the spot-overview map (Fig. 1c). Each module is characterized in terms of the list of genes included, their mean expression profile in all samples studied and a list of enriched gene sets enabling functional interpretation (Fig. 1d). Sample diversity plots, e.g. based on correlation network and correlation spanning tree algorithms visualize multivariate similarity relations between the samples (Fig. 1e, f). They support our definition of the molecular subtypes by forming well separated sample clusters.
A second use case addressing the expression landscapes of human tissues can be found in the supplement. It illustrates advantages of oposSOM data portraying compared to a ‘traditional’ two-way clustering heatmap.
4 Conclusion
oposSOM bundles a series of sophisticated analysis methods with intuitive visualization options to study high-dimensional data with the special focus on gene-centered expression data. It is designed for a broad user community ranging from bioinformaticians with demands for comprehensive analyses in a sophisticated workflow to application-oriented experimenters with needs in intuitive visualization options for their data.
Acknowledgements
This publication is supported by the Federal Ministry of Education and Research (BMBF), project grant No. FKZ 031 6166 (MMML-MYC-SYS) and FKZ 031 6065A (HNPCC-SYS).
Conflict of Interest: none declared.
References
Author notes
Associate Editor: Ziv Bar-Joseph


{kind=link}