





Abstract
Motivation: CYP2D6 is highly polymorphic gene which encodes the (CYP2D6) enzyme, involved in the metabolism of 20–25% of all clinically prescribed drugs and other xenobiotics in the human body. CYP2D6 genotyping is recommended prior to treatment decisions involving one or more of the numerous drugs sensitive to CYP2D6 allelic composition. In this context, high-throughput sequencing (HTS) technologies provide a promising time-efficient and cost-effective alternative to currently used genotyping techniques. To achieve accurate interpretation of HTS data, however, one needs to overcome several obstacles such as high sequence similarity and genetic recombinations between CYP2D6 and evolutionarily related pseudogenes CYP2D7 and CYP2D8, high copy number variation among individuals and short read lengths generated by HTS technologies.
Results: In this work, we present the first algorithm to computationally infer CYP2D6 genotype at basepair resolution from HTS data. Our algorithm is able to resolve complex genotypes, including alleles that are the products of duplication, deletion and fusion events involving CYP2D6 and its evolutionarily related cousin CYP2D7. Through extensive experiments using simulated and real datasets, we show that our algorithm accurately solves this important problem with potential clinical implications.
Availability and implementation: Cypiripi is available at http://sfu-compbio.github.io/cypiripi.
Contact: [email protected].
1 Introduction
Response to a large number of clinically prescribed drugs varies significantly among individuals. Although some patients show a good response to a medication, the same treatment might fail in others or cause serious side effects, which can even result in the death of the patient (Ma and Lu, 2011). In many cases, an individual’s genetic makeup has been recognized as one of the potential causes of treatment failures (Green et al., 2013). To avoid adverse effects, it is recommended to perform accurate genotyping prior to treatment decisions that include drugs sensitive to the allelic composition of genes involved in their metabolism (Cavallari, 2012). Drug dosage and selection can then be adjusted based on the inferred genotypes.
Cytochrome P450 2D6 (CYP2D6) is one of the most widely studied genes for which the correlation between the allelic makeup and therapy response has been established. It is currently estimated that metabolism of 20–25% of clinically prescribed drugs is, at least in part, dependent on CYP2D6 genotype (Ingelman-Sundberg, 2004). These include antidepressants, antipsychotics, anticancer drugs, opioids and many others (Zhou, 2009; Ingelman-Sundberg, 2004).
CYP2D6 is highly polymorphic gene with more than 100 different allelic variants reported up to date. The information about the known alleles is publicly available at CYP2D6 allele nomenclature website (http://www.cypalleles.ki.se/cyp2d6.htm), which contains detailed information on the sequence variants characterizing each allele. The website also includes information on the impact of genotype on the activity of the encoded enzyme for more than 50 known alleles. Based on their enzyme activity, the set of known CYP2D6 alleles is divided into four categories: poor, intermediate, extensive and ultrarapid metabolizers corresponding to ‘none’, ‘decreased’, ‘normal’ and ‘ultrarapid’ activity, respectively (Gaedigk et al., 2007). As genotyping techniques improve and more studies including large cohorts of individuals with different ethnic backgrounds are conducted, the existing database will expand to include novel alleles and more detailed, more accurate information on genotype–phenotype associations for known alleles.
Most of the known CYP2D6 variants are characterized by single-nucleotide polymorphisms (SNPs) and short insertions/deletions (indels). However, in addition to CYP2D6, the human CYP2D locus contains two pseudogenes CYP2D7 and CYP2D8, closely located and evolutionarily related to CYP2D6 (Kimura et al., 1989). The presence of highly homologous gene units in CYP2D6 and CYP2D7 facilitates crossing-over and formation of large gene conversions, deletions, duplications and multiplications (Kramer et al., 2009). Figure 1 depicts all of the known CYP2D gene arrangements.
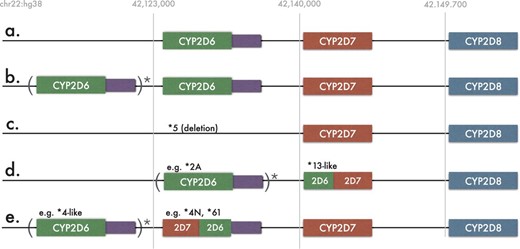
Five known CYP2D6 gene arrangements. The reference strand of human genome was used in all cases. Various number, including zero, of CYP2D6 copies is allowed within the parenthesis. (a) CYP2D6 non-duplicated arrangement consisting one copy of each of CYP2D6, CYP2D7 and CYP2D8. Purple rectangle represents CYP2D6 untranslated region. This region contains several variations important for the detection of some CYP2D6 alleles; (b) typical CYP2D6 duplication arrangement; (c) the deletion arrangement, indicating the absence of CYP2D6 (denoted as *5 allele); (d) CYP2D6/2D7 fusions (*13 family of alleles) lacking CYP2D7. Variable number of copies of CYP2D6 gene might precede fusion alleles; (e) CYP2D7/2D6 fusion cases with presence of CYP2D7. Variable number of copies of CYP2D6 gene might precede fusion alleles in this case as well
CYP2D6 also exhibits extensive copy number variation. Although the gene might be completely absent in some individuals, others who carry as many as 14 copies have been discovered (Ingelman-Sundberg, 2004).
Because of its clinical significance and the prevalence of genotypes resulting in altered phenotypes, several CYP2D6 genotyping platforms have been introduced. These usually include allele-specific primer extension assays, liquid bead arrays and TaqMan genotyping assays. However, several discrepancies among genotypes produced by these platforms have been reported (Pratt et al., 2010; Fang et al., 2014). Also, discoveries of some of the novel alleles and variations necessitate the extension of existing kits by construction and addition of novel primers, thereby increasing the time and cost required for genotype inference. The sensitivity of primers to sequence variation in primer binding sites can result in incorrect genotype assignment as described in Gaedigk et al. (2010). Furthermore, some of the techniques are incapable of detecting several alleles (Fang et al., 2014); they can also produce ambiguous readouts or incorrect estimates for individuals carrying hybrid genes (Gaedigk et al., 2010; Kramer et al., 2009). Another issue with some of the available approaches is their inability to differentiate between duplicated and non-duplicated alleles in samples with a duplication signal and heterozygosity (Kramer et al., 2009).
Recently introduced high-throughput sequencing (HTS) technologies represent a promising, time-efficient, cost-effective and potentially high-accuracy alternative to currently used genotyping techniques. In a single machine run, a typical HTS sequencing platform, like Illumina HiSeq 2000, generates billions of short DNA fragments/reads. Although these reads are substantially shorter than those generated by Sanger sequencing (75–250 bp versus 650–800 bp), their higher coverage provides improved indel and SNP detection accuracy. In addition, because leading HTS platforms (in particular Illumina) provide uniform sequence coverage, the copy number of a genomic region of interest can be estimated by comparing the expected and observed coverage in a given genomic region. Furthermore, the use of paired end reads can facilitate fine-grained inference of the origin of sequence variants commonly observed in both CYP2D6 and CYP2D7.
Despite rapid advances in HTS technologies, no available computational tool is capable of resolving the CYP2D6 genotype. A computational tool to solve this important problem needs to address many obstacles emerging from extensive allelic variation and sequence similarity between genes present at the CYP2D locus. Although this locus is unique in the human genome, the high degree of similarity among CYP2D genes results in an abundance of reads with multiple mapping locations. This can significantly complicate copy number analysis and accurate genotyping. As a large number of SNPs and indels that define some of CYP2D6 alleles can also occur in the pseudogene CYP2D7, detailed analysis of obtained variation signals is necessary. Failing to do so might result in inaccurate CYP2D6 genotype assignment caused by improper interpretation of variations originating from CYP2D7, mistakenly assigned to CYP2D6.
In this work, we present Cypiripi, the first algorithm for automatic CYP2D6 genotype inference from genomic HTS data. Cypiripi is able to properly resolve complicated configurations, including fusions between CYP2D6 and CYP2D7 genes, as well as both CYP2D6 and CYP2D7 deletions and duplications. We demonstrate that Cypiripi is highly accurate through extensive experiments involving both simulated and real datasets.
2 Methods
Cypiripi consists of the following main steps (Fig. 2):
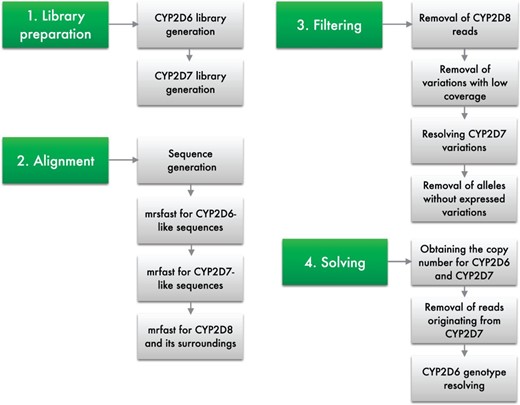
Graphical representation of the steps employed by our framework
Library preparation step, where a library containing the complete set of relevant variations occurring in CYP2D locus is constructed.
Read alignment step, where each HTS read is aligned to the library of gene variants from the CYP2D locus determined in the library preparation step.
Filtering step, where alleles with sequence variations that lack appropriate read support are removed from further consideration.
Combinatorial optimization step, where the genotype, consisting of the composition of CYP2D6 allelic variants and their copy numbers, is inferred by using Integer Linear Programming (ILP).
2.1 Library preparation
In this step, we construct a library containing the information about currently available variations occurring within CYP2D locus. These include SNPs, indels and details about recombination events occurring between CYP2D6 and CYP2D7. Because of the nature of the problem being solved, we mainly focus on variations that define the currently known CYP2D6 alleles.
Variations occurring in CYP2D6 have been extracted from the most recent update (December 2014) of the database at the CYP2D6 allele nomenclature website. The corresponding information is stored in the simple text file and any subsequent changes in on-line database can be easily incorporated in our tool by a straightforward modification of this file.
CYP2D7 library reconstruction is harder due to the fact that there is no basepair level characterization available for CYP2D7 alleles. To be able to differentiate CYP2D7 from CYP2D6, we found 10 available sequences from GenBank and other sources (see the Appendix A) and aligned them with Clustal (Larkin et al., 2007) to obtain a consensus alignment for CYP2D7. This consensus was aligned to the CYP2D6 reference allele, and the set of differences between those two consensus sequences were used as markers for identifying CYP2D7 presence and for generating CYP2D7 reference sequence. These markers were also used for proper description of the fusion and conservation alleles (i.e. alleles involving a portion—e.g. a whole exon—of a CYP2D6 allele, swapped with the similar sequence portion of a CYP2D7 allele). Note that those markers are not intended to be authoritative, since there might exist uncatalogued CYP2D7 alleles which do not contain any of those markers. Our formulation takes that into consideration and uses only markers which have sufficient support to infer the presence of CYP2D7 (e.g. insertion of T at loci 137 indicates with high probability that CYP2D7 region is present).
Our method does not require a database with CYP2D8 variants for the reasons described below.
2.2 Read alignment
We established the uniqueness of CYP2D gene sequences by searching for the entire human genome (excluding the CYP2D locus) regions with high sequence similarity to the CYP2D genes by BLAT (Kent, 2002). No subsequence of length >60 bp from any one of the CYP2D genes can be found in the remainder of the human genome within an edit distance of 6.
As the length of the reads generated by current HTS technologies usually exceeds 60 bp, each of the reads that can be aligned (with only a few differences) to CYP2D6 genes must originate from the CYP2D cluster. Thus, to extract the reads originating from CYP2D genes, it is sufficient to map all of the reads against this set and discard those that cannot be aligned successfully. This alignment is performed by our in-house developed mrfast and mrsfast family of multi-mapping tools (Alkan et al., 2009; Hach et al., 2010, 2014). For each of the successfully aligned reads, we keep the details about the genes and locations it can be aligned to and variants in the library it supports. Alignment details, respectively, for CYP2D6, CYP2D7 and CYP2D8 are given below.
To perform an alignment of the reads against all possible CYP2D6 alleles, we constructed each allele at basepair resolution. For that we combined information from CYP2D6 reference sequence M33388 (http://www.ncbi.nlm.nih.gov/nuccore/181303) and CYP2D6 variant database we constructed in the previous step. The alignment was then performed using mrsfast, allowing at most two mismatches per read.
Because of the lack of a comprehensive list of sequence variants commonly observed in CYP2D7, there could be SNVs or short indels not represented in the consensus sequence we constructed. To account for such variants and possible sequencing errors, we set the maximum number of errors (mismatches and indels - i.e. edit distance) to 5. The alignment of reads was performed by mrfast, which allows mismatches and indels. Since reads originating outside of the CYP2D locus have an edit distance more than 5 to any CYP2D gene, any read aligning with the consensus sequence should be originating from the CYP2D locus.
Although CYP2D8 is evolutionarily related to CYP2D6 and CYP2D7, its sequence composition is significantly different from that of the other two. In addition, there are no recombination events involving CYP2D6 and CYP2D8. As a result, we assume that CYP2D8 is always located downstream of CYP2D7 (considering 5’–3’ orientation in the human reference genome) and that, the vast majority of the reads originating from this gene are not mappable to the other two genes. However, there are two 0.5 kb flanking regions at CYP2D8 boundaries that can give rise to some reads mappable to CYP2D6 and/or CYP2D7 (Fig. 3 ). Such reads can interfere with copy number and other key estimates for CYP2D6 and CYP2D7. It is therefore important to filter out such reads. We thus perform an alignment of the reads against CYP2D8 gene and its surroundings by using the CYP2D8P reference sequence M33387 (http://www.ncbi.nlm.nih.gov/nuccore/M33387) by the use of mrfast, with edit distance 5. Reads falling into the abovementioned flanking regions around CYP2D8 are marked for filtering, as described in the next step.

The coverage of the reads mappable to the CYP2D6 and/or CYP2D7 genes is depicted in grey on the flanking regions of CYP2D8 (blue strip). Only two small 0.5 KB regions on the sides accept CYP2D6 and/or CYP2D7 reads
2.3 Filtering
After the first two steps, we obtain a set of reads mappable to at least one of the CYP2D genes together with the details about the exact locations they align to and the sequence variants they support. In addition, for each variant, we have information about the number of reads supporting it. To remove false positives and lower the search space for the combinatorial optimization step, we perform several read, variation and allele filtering steps with the details below.
2.3.1 CYP2D8 read filtering
Since we know the regions in the CYP2D6 and CYP2D7 to which some of CYP2D8 reads can map to, we can use the surroundings of these regions to find out the excess coverage generated by such reads. After finding excess coverage, we remove the reads to ‘flatten’ the coverage of the region with its surroundings.
2.3.2 Variation filtering
Sequencing errors can result in support for sequence variants that do not exist in the underlying genome. For example, assume that our library contains the SNP G > A at genomic position p. Also, assume that the underlying genome does not contain this SNP. In principle, a sequencing error occurring at position p may result in some support for nucleotide A instead of G. Because of the low sequencing error in the data we use, it is very unlikely that read support for non-existing variants will be significant. We therefore filter out all potential sequence variants that have support lower than user-specified parameter η.
2.3.3 CYP2D7 variation filtering
It is of great importance for our method to detect all variation coming from the CYP2D7 reads, which are falsely aligned to some of the CYP2D6 alleles. This is particularly important due to the fact that many key sequence variants used for CYP2D6 allele identification might also occur in CYP2D7. For example, c.1661 G > C is commonly found both in CYP2D7 and many of the CYP2D6 alleles. It is usually not clear whether this variant is associated with CYP2D7 or CYP2D6 or both. Commonly, CYP2D7-specific variants are found within the close vicinity (usually within the 100 bp) of the shared variants. This helps with the detection of the origin of shared variations by using only read alignment information. Unfortunately, in some cases, CYP2D7-specific variants are not present in the vicinity of shared variants (within a distance comparable to the read length). Unresolved shared variants can falsely indicate the existence of specific CYP2D6 alleles, which, in reality may not be present in the sequenced genome. For example, c.3853 G > A is shared by both CYP2D6*27 and CYP2D7; the closest CYP2D7-specific variation is more than 100 bp away from this locus. Relying solely on this information, we cannot decide whether it is *27 or *1 (the wild type allele) together with a CYP2D7 harbouring this variation, that is present. Fortunately, this problem can be resolved through the use of paired-end sequencing with a fragment length of 300 bp or more, whose span would help detect CYP2D7-specific variants.
2.3.4 Allele filtering
About 60% of the CYP2D6 alleles from the database are easily distinguishable from other alleles by at least one unique variant in their characterization. Each CYP2D6 allele whose unique variants are not supported after previous filtering steps are removed from further consideration. Unfortunately, the absence of a comprehensive list of CYP2D7 variants prevents us from applying this stringent filtration rule to the CYP2D7 gene.
2.4 Combinatorial optimization
The goal of the combinatorial optimization step is to find a genotype which best describes the set of reads remaining after previous read filtering steps. The optimal genotype is supposed to match the observed read coverage as closely as possible, as explained below.
2.4.1 Notation
Let L denote the set of variants from CYP2D6 and CYP2D7 variation library that have non-zero read support after previous filtering steps.
Consider an arbitrary variant . Assume that w starts at position j in the CYP2D6 reference sequence. In addition to the reads supporting w, there might also exist some reads spanning location j and not supporting any variation from L at location j. Since our optimization step also requires the number of such reads to be available, to detect the wild-type (*1) and other alleles without any variation at location j, we introduce the notion of a neutral SNP variation denoted by n(w), defined as the special type of ‘variation’ that preserves the reference nucleotide at location j. To illustrate this, consider the following example where w denotes the c.1661 G > C in CYP2D6. As this SNP starts at position 1661, the corresponding n(w) in this case is defined to be c.1661 G > G. Neutral SNP variation n(w) is harboured by all alleles that do not harbour any variation starting at j.
Let coverage of be the number of unfiltered reads supporting v and be denoted by .
Let Vi denote the set of variations defining the i-th allele. Clearly .
2.4.2 Formulation
The problem is formulated as an instance of ILP and solved using IBM CPLEX optimization software. To formulate the problem, we use the assumption that the average coverage of the HTS experiment is uniform and that its value is the user-provided parameter λ.
Define ai as an integer variable denoting the number of copies of the i-th CYP2D6 allele in the given sample. Let , where N denotes the number of different alleles. We assume that the total number of copies of CYP2D6 is upper bounded by a given parameter c. In this study, we set c = 20 which is greater than the maximum number of CYP2D copies found so far in a single individual (see Section 1). To incorporate this into our ILP, we add the constraint for each i.
We use a two-stage approach to solve the genotyping problem. In the first stage, we use previously described ILP formulation to obtain the copy number for CYP2D6 and CYP2D7, without making any decision about CYP2D6 genotypes. On the basis of this, we can remove the CYP2D7 reads and estimate the exact coverage at each location. Since the removal of CYP2D7 reads also removes the support for many shared variations, we perform an additional round of filtering to further reduce the number of potential false positives. Finally, we invoke a slightly modified version of the abovementioned ILP formulation to detect specific CYP2D6 genotypes, as described below.
After the optimal solution for this extended ILP is found, we report the final genotype consisting of all alleles i such that ai > 0 in the optimal solution. The copy number of each included allele i is set to ai.
Although they are extremely unlikely, there are very few cases where discerning between different CYP2D6 genotypes is theoretically infeasible using typical HTS data. In these cases, the proposed ILP has more than one optimal solution resulting in at least two different but equally likely genotypes. These cases occur when there are two sets of alleles, denoted A1 and A2, satisfying the following conditions:
The union of variations defining alleles from A1 is identical to the union of variations defining alleles from A2.
Ambiguous variations from those sets are not close enough to be covered by the paired-end reads originating from only one allele.
One simple example for this is shown in Figure 4 . In this figure, the presence of depicted SNPs can signal the genotype combination of either *34/*6 C or *2D/*6B. As key SNPs for discerning these two possibilities are more than 1000 bp apart, we cannot resolve this ambiguity using typical paired-end read data.
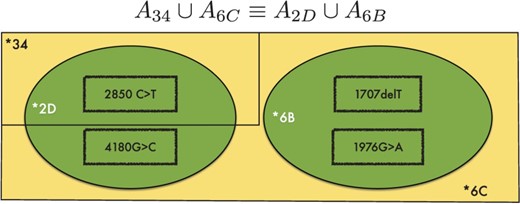
The ambiguous case where two genotypes *6C/*34 (yellow) and *2D/*6B (green) are equally likely. Key c.2850 C > T and c.4180 G > C are too distant to be resolved with the currently available HTS data
In such cases, we report all of the most likely genotypes together with a warning that there is ambiguity in the inferred genotype. If these genotypes result in different phenotypes, further sample analysis is required. With the help of upcoming advances in HTS technologies and the increase in read lengths and insert sizes, we expect to resolve this problem in the near future.
3 Results
The first set of validated CYP2D6 genotypes have recently been made available (Fang et al., 2014) for publicly available HTS data from 1000 Genomes Project Phase I collection (1000 Genomes Project Consortium, 2012). Unfortunately, none of these samples are suitable for our purposes as they are either sequenced at very low coverage (2–5x) or have very short read length (36 bp). Our method requires a minimum coverage of 10x per strand to successfully filter out the noise originating from sequencing errors. Furthermore, reads longer than 60 bp can ensure unambiguous mapping, as described in the filtering step of the Section 2. As a result, proper CYP2D6 genotype data for publicly available HTS experiments with reasonable coverage and read length is not yet available.
To evaluate the performance of Cypiripi, we custom designed benchmark data consisting of the following:
Simulation data: Cypiripi was evaluated on 71 simulated datasets designed to reflect known CYP2D6 genotypes (Kramer et al., 2009), including theoretically possible but highly unlikely cases;
Real data: Cypiripi was evaluated on publicly available CEPH 1463 trio (mother, father and son) sequenced by Illumina HiSeq 2000 platform with average coverage of 100 x per chromosome.
3.1 Simulations
Five sets of simulations, each covering a unique class of CYP2D6 allelic arrangement, were created for evaluating the performance of Cypiripi. Those arrangements were constructed with the aim of covering all possible allelic combinations, including copy number changes and fusion events, as depicted in Figure 1. Within each set, we simulated several individuals with the set’s specific allelic arrangement. The sets are defined as follows:
Diploid case where both maternal (M) and paternal (P) chromosomes have the allele of the same type (e.g. *1/*1).
Diploid case where both chromosomes contain one allele each and the alleles are different (e.g. *1/*3A).
Both chromosomes contain a common tandem duplication or deletion event (e.g. 5 ×*2X/5 ×*2X or *5/*5); note that *5 allele describes a CYP2D6 deletion.
Both chromosomes contain a common variety of different alleles (e.g. *1E *14B *2X *14A for every chromosome).
Both chromosomes contain a CYP2D7 fusion or conservation event (e.g. *13A/*13A or *4A *68A/*4A *68A).
Note that Cypiripi reports the total number of alleles found in an individual genome without making distinction between chromosomes (e.g. *1/*1 will be reported as 2 ×*1).
Also note that set (d) is quite unrealistic, since such cases with large number of distinct variants are yet to be observed. We include these samples to show the generality of the method and to evaluate its ability to cope with complex cases which could be encountered in the future. Sets (c) and (e), on the other hand, were specifically designed to reflect some of the previously discovered and validated genotypes (Kramer et al., 2009).
For every sample, we separately constructed the sequences of maternal and paternal chromosomes, based on chromosome 22 of human reference genome, version hg38. We have inserted in each chromosome the corresponding CYP2D6 gene within the coordinates of chr22:42 122 966–42 132 410. We have also replaced CYP2D7 with some of the randomly selected CYP2D7 genes mentioned in the Appendix A, to account for CYP2D7 variability between different individuals. In the case of fusions and duplications, we have followed the guide from Kramer et al. (2009), as depicted in Figure 1.
Simulated reads were generated by using simNGS (http://www.ebi.ac.uk/goldman-srv/simNGS/) simulator, which is capable of accurately simulating Illumina HiSeq 2000 machine parameters (details in the Appendix B), including substitution and indel rate. We have generated 101-bp paired-end library with the average insert size of 400. Paired-end coverage per chromosome was around 20x, totalling average coverage of 40x per individual. Although the current standard is approaching 200x per individual, we opted for lower coverage to show the robustness of the method.
All simulation results are listed in Table 1 . Cypiripi performed extremely well, providing 100% correct genotype for majority of the cases (62 out of 71). In four out of nine remaining cases, Cypiripi reports an allele belonging to the same family as the correct allele (e.g. *4E and *4C from sample 33 belong to the same family *4). Copy number estimation was not in agreement with the ground truth in only two cases (samples 51 and 58), both having very large CYP2D6 copy number (16 and 14, respectively). In these two cases, the inferred copy number was lower by one compared with the ground truth. In all other cases, copy number was identified properly. Sample 26 contains ambiguous genotype described in Section 2, and in this case, Cypiripi reported both genotypes as equally likely.
Cypiripi performance for every group
 |
|
Correctly identified alleles are shown in green, while incorrect estimates are reported in red colour. In case of mismatches, red colour is also applied to the second column items to pinpoint the problematic allele. Unless otherwise specified, genotypes are given for both maternal and paternal chromosome in the format M/P. For set (d), where both chromosomes have the same allelic combination, we only show content of one chromosome for the sake of brevity. Results for set (d) are still reported for each chromosome separately. For ambiguous cases, all optimal genotypes are reported (e.g. 26th sample).
Cypiripi performance for every group
|
|
Correctly identified alleles are shown in green, while incorrect estimates are reported in red colour. In case of mismatches, red colour is also applied to the second column items to pinpoint the problematic allele. Unless otherwise specified, genotypes are given for both maternal and paternal chromosome in the format M/P. For set (d), where both chromosomes have the same allelic combination, we only show content of one chromosome for the sake of brevity. Results for set (d) are still reported for each chromosome separately. For ambiguous cases, all optimal genotypes are reported (e.g. 26th sample).
All samples from Table 1 contained two copies of CYP2D7 (one for maternal and for paternal chromosome), excluding the samples containing *13 allele (because all *13 fusions imply the removal of CYP2D7). The number of CYP2D7 genes was estimated correctly in all samples.
Cypiripi has a special mode to detect and resolve fusion cases. The main difference consists of less stringent filtering used for samples containing fusions and conservations, since such alleles contain the same set of uncertain variations as CYP2D7. It is important to stress, as can be seen from the set (e) in the Table 1, that Cypiripi is able to successfully handle various fusion cases. The only problematic case is misdetection of *13F and *13H as CYP2D7. Unfortunately, these fusions occur at the end of exon 9, preserving majority of CYP2D7 and just a small portion of CYP2D6*1. As all CYP2D7-specific variations are present in *13F and *13H, Cypiripi might detect either CYP2D7 or *13F/H. Because of the fact that all *13 alleles encode the poor metabolizer as does CYP2D7, the corresponding phenotype is still accurately assigned based on the reported genotype.
We set the parameter η to be . Higher values perform better when the copy number is very high. Thus, we used for the set (c).
It is worth mentioning that Cypiripi is a highly optimized and efficient tool. It requires only few minutes for a simple sample with two CYP2D6 copies and no more than 10 min on any other sample we evaluated on Intel Xeon 3.50 GHz CPU. This makes it ideal choice for clinical environments where the speed is of high importance.
3.2 Real data
To evaluate the performance of Cypiripi on real data sets, we used the family trio from CEPH 1463 pedigree. This trio consists of mother, father and son with high-coverage Illumina HiSeq 2000 sequencing data publicly available for each of its members (http://www.illumina.com/platinumgenomes/). In addition to the sequencing data, in their recent article, Zook et al. (2014) identified the highly confident SNPs for NA12878 (mother) which belongs to this trio. The analysis of these SNPs confirmed the presence of two CYP2D6 copies. The first copy was validated as CYP2D6*3 A and the obtained signal allows for the validation of second copy up to the allelic family level (CYP2D6*4). A genotype inferred by Cypiripi is in the agreement with both of these results. Namely, it was able to accurately identify the existence of CYP2D6*3A and reported the second copy as CYP2D6*4M.
Cypiripi reported *4M/*4M as a genotype for both father and son (Table 2 ). Although we do not have ground truth about CYP2D6 genotypes for these two individuals, these predictions are in the strong agreement with Mendelian laws of inheritance.
Cypiripi predictions for real data set
| CEPH 1463 trio dataset | |
|---|---|
| ID | Identified |
| NA12877 (father) | *4M/*4M |
| NA12878 (mother) | *3A/*4M |
| NA12882 (son) | *4M/*4M |
| CEPH 1463 trio dataset | |
|---|---|
| ID | Identified |
| NA12877 (father) | *4M/*4M |
| NA12878 (mother) | *3A/*4M |
| NA12882 (son) | *4M/*4M |
NA12878 predictions are coloured in green due to the fact that they match the highly confident SNP calls from Zook et al. (2014). Since the validated predictions are not available for the other two samples, predictions of their genotypes are coloured black.
Cypiripi predictions for real data set
| CEPH 1463 trio dataset | |
|---|---|
| ID | Identified |
| NA12877 (father) | *4M/*4M |
| NA12878 (mother) | *3A/*4M |
| NA12882 (son) | *4M/*4M |
| CEPH 1463 trio dataset | |
|---|---|
| ID | Identified |
| NA12877 (father) | *4M/*4M |
| NA12878 (mother) | *3A/*4M |
| NA12882 (son) | *4M/*4M |
NA12878 predictions are coloured in green due to the fact that they match the highly confident SNP calls from Zook et al. (2014). Since the validated predictions are not available for the other two samples, predictions of their genotypes are coloured black.
The coverage parameter λ for those samples was set to 100, with the exception of NA12877, whose measured coverage was lower and was equalling 90. The η was, as it was the case with the simulated samples, set to .
4 Conclusion
In this article, we have presented the first computational framework to exactly characterize the clinically important CYP2D6 gene and its variations by using HTS data only. Our framework, which we call Cypiripi, is able to cope with many of the issues presented by the existing (non HTS based) approaches for CYP2D6 genotyping, such as their inability to perform accurate copy number estimation, CYP2D7 variant characterization and fusion detection.
In addition, Cypiripi’s highly optimized running time makes it an ideal choice for clinical settings where speed is of high importance. The algorithmic basis of Cypiripi, a gene-agnostic ILP, can be easily extended to other unique gene clusters with similar properties.
It should be noted that there remain some challenges that we aim to investigate in follow-up work. For example, genotyping when the available set of sequence variants can be described by more than one set of genotypes is problematic. This technology-bound issue can be resolved by the use of paired end reads in some cases but may require the availability of longer reads for the resolution of other cases. In addition, exact characterization of novel genotypes within the CYP2D locus is a further goal to be investigated.
As the cost of whole-genome sequencing (WGS) plummets and approaches the cost of exome sequencing, we will be able to perform detailed sequence analysis of several clinically important loci across the human genome by using standard coverage HTS data. This can reduce both the time and cost required for genomic analysis and address many of the limitations of existing (non-HTS-based) techniques.
As WGS makes its way into the clinic, it is providing economical and efficient means to identify many pharmacogenomic variants that can be used to provide personalized medication options. By the use of a proper computational framework such as Cypiripi, decision support systems to assist physicians for prescribing specific medications can benefit from fast and accurate genotyping based on HTS.
Acknowledgement
We thank F. Hach, E. Hodžić and C. Koçkan for proof reading and suggestions during the preparation of the manuscript.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Frontiers project, "The Cancer Genome Collaboratory" as well as NSERC Discovery Grants program and Genome Canada (to S.C.S.), the Vanier Canada Graduate Scholarship program (to I.N.) and NSERC Create (to S.M.), NIH R01GM088076 (T.C.S), and the NIH IGNITE project grant (U01HG007762) (D.A.F., T.C.S., V.M.P.). This publication was made possible by the Indiana University Health Indiana University School of Medicine Strategic Research Initiative (to V.M.P.).
Conflict of Interest: none declared.
References
Appendix A: CYP2D7 variations
The CYP2D7 genes were extracted from the following sequences:
a) M33387
b) NW_003315971.2
c) NT_187682.1
d) NC_000022.11
e) AC_000154.1
f) NC_018933.2
g) ENSG00000205702.2
(http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000205702)
h) hg19 reference genome
(chr22:42536214–42540575)
i) hg38 reference genome
(chr22:42140203–42144549)
j) NA12878 Assembly, Maternal Chromosome
(22:42534697–42539033)
h) NA12878 Assembly, Paternal Chromosome
(22:42534225–42538562)
The NA12878 assembly was accessed from http://sv.gersteinlab.org/NA12878_diploid/.
Appendix B: program parameters
simNGS was invoked as:
simLibrary -x <coverage> <fasta> |
simNGS -o fastq -p paired <runfile>> <fastq>
The Illumina HiSeq runfile for simNGS was obtained from http://www.ebi.ac.uk/goldman-srv/simNGS/runfiles/101cycleHiSeq/s_3_4x.runfile.
Cypiripi was invoked as:
cypiripi -C <coverage> -T <eta> -r <library> -s <mapping.sam>
Fusion datasets were run with the addition of -F parameter.
Author notes
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}