





Abstract
Summary: Protein-coding genes with multiple alternative polyadenylation sites can generate mRNA 3′UTR sequences of different lengths, thereby causing the loss or gain of regulatory elements, which can affect stability, localization and translation efficiency. 3USS is a web-server developed with the aim of giving experimentalists the possibility to automatically identify alternative 3 ′ UTRs (shorter or longer with respect to a reference transcriptome), an option that is not available in standard RNA-seq data analysis procedures. The tool reports as putative novel the 3 ′ UTRs not annotated in available databases. Furthermore, if data from two related samples are uploaded, common and specific alternative 3 ′ UTRs are identified and reported by the server.
Availability and implementation: 3USS is freely available at http://www.biocomputing.it/3uss_server
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The 3 ′ untranslated regions (3 ′ UTRs) of mRNAs play a crucial role in post-transcriptional regulation. In fact, they contain cis- regulatory elements, such as AU-rich elements (ARE), microRNA binding sites, etc. ( Khabar, 2005 ; Bartel, 2009 ), which can affect transcript stability, degradation, subcellular localization and translation. Through alternative polyadenylation, a widespread mechanism in eukaryotic organisms, different isoforms of the same gene can acquire alternative (longer or shorter) 3 ′ untranslated regions ( Elkon et al ., 2013 ). The phenomenon seems to be different in different tissues ( Lianoglou et al ., 2013 ), during development ( Ji et al ., 2009 ) and, recently, it has also been observed in cancer cells ( Mayr and Bartel 2009 ). With the emergence of next-generation sequencing technologies, it is now possible to sequence entire transcriptomes in one experiment. In standard RNA-seq data analysis protocols the reconstructed transcripts are compared with an annotated Reference transcriptome. The most commonly used methods [e.g. Cuffcompare and Cuffmerge ( Trapnell et al ., 2012 )] distinguish known from putative novel isoforms, but they rely on the comparison of the intronic structure of the transcripts, thereby do not take into account reads that might support the existence of alternative 3 ′ UTRs and therefore the existence of putative novel transcripts with different 3 ′ UTR is not reported to the user. The great interest for these important regulatory regions is leading to the development of more sophisticated RNA sequencing library preparation protocols (the more powerful will be the possibility to entirely sequence the untranslated region, the more accurate will the reconstruction of putative novel 3'UTRs be). At the same time, different computational tools to capture [ Lu et al ., 2013 ; Wang et al ., 2014 ; github.com/vodkatad/roar (R-package)] and store (compbio.dundee.ac.uk/polyADB/, mosas.sysu.edu.cn/utr) the 3 ′ ends of polyadenylated transcripts have been recently developed. Nevertheless, the identification of 3 ′ UTR alternative regions still remains a challenge. 3USS (3 ′ UTR Sequence Seeker) is the only available web-server developed to automatically identify the assembled transcripts with alternative 3 ′ UTRs with respect to the ones in Reference transcriptomes and provides the user with their nucleotide sequence together with their genomic coordinates and other information, such as their difference in length and the corresponding gene. Furthermore, when data from two related experiments are provided, the reconstructed 3 ′ UTRs of each biological sample are identified and compared.
2 Description
The server starts the analysis from a transcriptome assembly file obtained by standard computational methods for RNA-seq transcript reconstruction, such as Cufflinks ( Trapnell et al ., 2012 ), Scripture ( Guttman et al ., 2010 ) and others. Indeed, the 3USS procedure is independent from the transcriptome reconstruction method used to obtain the RNA-seq assembly file as long as the data have been compared with a Reference annotation [such as UCSC ( Rhead et al . 2010 ), NCBI ( Pruitt et al ., 2014 ), Ensembl ( Flicek et al ., 2010 ), Gencode ( Harrow et al ., 2012 ), etc.] using Cuffcompare or Cuffmerge (see the Help page for full information). 3USS also permits to easily retrieve the genomic coordinates and sequences of transcripts already annotated in public databases.
2.1 Input
For identifying RNA-seq assembled transcripts with alternative 3 ′ UTRs with respect to a Reference, the user needs to provide: the organism and related genome version; the Reference annotation transcriptome and one or two RNA-seq transcriptome assembly files (as above described). An example using public data from GEO database ( Barrett et al . 2013 ) is accessible from the 3USS input page.
2.2 Results
Each assembled transcript is processed by selecting the isoforms sharing the same intron chain of known protein-coding transcripts in the Reference. The reconstructed 3 ′ UTRs are consequently identified as the regions located immediately after the stop-codon and can be directly compared, detecting possible differences in their 3 ′ UTR lengths. The alternative 3 ′ UTRs are then compared with already annotated ones in other databases [the latest available data from iGenomes ( http://cufflinks.cbcb.umd.edu/igenomes.html ) and Gencode ( Harrow et al ., 2012 )] to identify putative novel (not yet annotated) 3 ′ UTRs. The most informative output files that the 3USS server produces are: a fasta file with the alternative 3 ′ UTR nucleotide sequences and other information in their headers (gene Id, coding strand, 3 ′ UTR genomic coordinates and length); a list of the transcripts with their 3 ′ UTR length difference and a list of the alternative not yet annotated 3 ′ UTRs (putative novel) in other databases as well as the annotated ones. The user can also easily visualize the transcripts in a graphical environment through the UCSC Genome browser ( Kent et al ., 2002 ; see examples in the Supplementary Material ). If an already annotated transcriptome is uploaded as input, the 3 ′ UTR sequences and genomic coordinates of all the protein-coding transcripts are returned. If the user submits input files derived from two different related RNA-seq experiments, the 3USS server compares the alternative 3 ′ UTRs, reporting how they are distributed across the two biological samples ( Fig. 1 ).
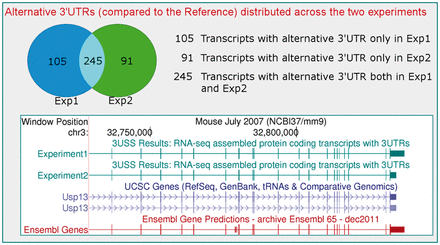
Some results in case of two RNA-seq experiments (see Help page and Suppl. mat. for full information). A Venn diagram illustrates how the transcripts with alternative 3 ′ UTRs are distributed across the two experiments. An example of the graphical view of the assembled transcripts (in dark green) with specific 3 ′ UTRs in different experiments is obtained through the links to the UCSC Genome Browser ( http://genome.ucsc.edu )
Each transcript with an alternative 3 ′ UTR is associated to its assembled transcript id (e.g. TCONS_0000X, for Cufflinks transcriptome assembly files), and through it, to all the corresponding analysis results, so that useful information (read coverage, expression abundance, etc.) can be directly retrieved from the complete RNA-seq data analysis results.
3 Conclusion
To the best of our knowledge, 3USS is the only available web-server that can automatically detect the presence of isoforms with alternative 3 ′ UTRs in the RNA-seq experiment under investigation and output their genomic coordinates and nucleotide sequences for further analysis. Moreover, we believe that a protocol independent tool, such as the one described here, is useful and not only to retrieve information on novel 3 ′ UTRs, but also to easily compare the results of different methods and approaches. Through an easy to use interface, it also allows the comparison of the 3 ′ UTRs of the sequenced transcripts in different biological conditions. This allows researchers to further examine genes affected by 3 ′ UTR shortening or lengthening and, for example identify their biological relevance using public tools such as FIDEA ( D'Andrea et al ., 2013 ) or others, as well as directly analyze the novel 3 ′ UTR nucleotide sequences to detect differences in the presence or absence of regulatory motifs and/or binding sites.
Acknowledgements
The authors thank the Biocomputing Unit for interesting discussions.
Funding
This project was supported by KAUST [Grant Number KUK-I1-012-43], PRIN 20108XYHJS and Epigenomics Flagship Project—EPIGEN. Funding for open access charge: KAUST.
Conflict of Interest : none declared.
References
Author notes
Associate Editor: John Hancock


{kind=link}