





Abstract
Motivation: Upstream open reading frames (uORFs) are often found in the 5′-untranslated regions of eukaryotic messenger RNAs. Some uORFs have been shown to encode functional peptides involved in the translational regulation of the downstream main ORFs. Comparative genomic approaches have been used in genome-wide searches for uORFs encoding bioactive peptides, and by comparing uORF sequences between a few selected species or among a small group of species, uORFs with conserved amino acid sequences (UCASs) have been identified in plants, mammals and insects. Regulatory regions within uORF-encoded peptides that are involved in translational control are typically 10–20 amino acids long. Detection of homology between such short regions largely depends on the selection of species for comparison. To maximize the chances of identifying UCASs with short conserved regions, we devised a novel algorithm for homology search among a large number of species and the automatic selection of uORFs conserved in a wide range of species.
Results: In this study, we developed the BAIUCAS (BLAST-based algorithm for identification of UCASs) method and identified 18 novel Arabidopsis uORFs whose amino acid sequences are conserved across diverse eudicot species, which include uORFs not found in previous comparative genomic studies due to low sequence conservation among species. Therefore, BAIUCAS is a powerful method for the identification of UCASs, and it is particularly useful for the detection of uORFs with a small number of conserved amino acid residues.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Upstream open reading frames (uORFs) are small ORFs located in the 5′-untranslated regions (5′-UTRs) of many eukaryotic mRNAs. Recent genome-wide analyses revealed that 10–50% of eukaryotic genes contain one or more uORFs (Churbanov et al., 2005; Galagan et al., 2005; Kawaguchi and Bailey-Serres, 2005; Rogozin et al., 2001). The presence of a uORF can modulate the translational efficiency of the downstream main ORF (mORF) (Calvo et al., 2009). Regulatory roles of uORFs have been demonstrated in processes such as the stress response and feedback regulation of biosynthesis (Calvo et al., 2009; Hood et al., 2009; Meijer and Thomas, 2002; Morris and Geballe, 2000; Vilela and McCarthy, 2003).
Although the effects of most uORFs seem to be independent of their amino acid sequence, several uORFs have been shown to affect the translation of the mORF in an amino acid sequence-dependent manner (Hanfrey et al., 2005; Ivanov et al., 2008; Jousse et al., 2001; Morris and Geballe, 2000; Rahmani et al., 2009). For example, in the translational feedback regulation of the Neurospora crassa arg-2 gene, which encodes the small subunit of arginine-specific carbamoyl phosphate synthetase, the 24-residue nascent peptide encoded by the arg-2 uORF causes ribosome stalling at the stop codon of the uORF in response to arginine, resulting in translational repression of the mORF (Gaba et al., 2001; Wang and Sachs, 1997). In plants, uORFs with conserved amino acid sequences (UCASs) have been reported to be involved in the translational regulation of four Arabidopsis thaliana genes (Hanfrey et al., 2005; Imai et al., 2006; Rahmani et al., 2009; Tabuchi et al., 2006), and the importance of uORF-encoded amino acid sequences has been demonstrated in the translational regulation of the SAMDC 1 and bZIP 11 genes (Hanfrey et al., 2005; Rahmani et al., 2009).
Although uORFs present only in a limited number of species can have a regulatory role, it is more likely that evolutionarily conserved uORFs have a regulatory function. Therefore, to identify uORFs with potential regulatory roles, genome-wide searches for conserved uORFs have been conducted using comparative genomic approaches in various organisms, including mammals (Churbanov et al., 2005; Iacono et al., 2005; Zhang and Dietrich, 2005), yeasts (Crowe et al., 2006; Cvijovic et al., 2007), plants (Hayden and Jorgensen, 2007; Tran et al., 2008), insects (Hayden and Bosco, 2008) and fungi (Neafsey and Galagan, 2007). Some of these studies searched for UCASs to identify peptide sequence-dependent regulatory uORFs. In plants, Hayden and Jorgensen identified 19 groups of UCASs by comparing uORF sequences between orthologous genes of A. thaliana and rice. Additionally, they found seven groups of UCASs by comparing A. thaliana paralogous genes. These UCASs were referred to as conserved peptide uORFs (CPUs) (Hayden and Jorgensen, 2007). In mammals, uORF sequences were compared between orthologous genes in human and mouse, and over 200 UCASs were identified (Crowe et al., 2006). In insects, 44 UCASs were identified by comparing uORF sequences among dipteran species (Hayden and Bosco, 2008). Thus, in these previous studies, uORF sequences were compared between a few species or among a small group of species.
In most bioactive uORF peptides that have been experimentally shown to function in translational control, the region involved in regulation is typically 10–20 amino acids long (Alderete et al., 1999; Ivanov et al., 2008; Morris and Geballe, 2000; Rahmani et al., 2009; Spevak et al., 2010). Therefore, it is important to detect homology between such short regions for a comprehensive identification of UCASs. However, if uORF sequences are compared between only a few selected species, the detection of homology between such short peptide sequences largely depends on the species selected for comparison. If the conservation of the uORF sequences among the selected species is insufficient to detect the similarity, then the uORF would not be identified even if the uORF sequences are sufficiently conserved among other species. If uORF sequences are compared between closely related species, the observed similarity between short uORF-peptide sequences may be due to nucleotide sequence retention rather than functional preservation of the peptides.
To overcome these problems that result from the selection of species for comparative genomic analyses, we developed a novel method, BAIUCAS (BLAST-based algorithm for identification of UCASs), in which homology searches are performed using an expressed sequence tag (EST) dataset derived from thousands of species, and uORFs conserved in a wide range of species are efficiently selected. Using this method, we identified 18 novel A. thaliana uORFs whose amino acid sequences are conserved across diverse plant species.
2 METHODS
2.1 BAIUCAS algorithm
We developed BAIUCAS to conduct an exhaustive search for UCASs. This algorithm consists of a six-step procedure (Fig. 1). The first step is an exhaustive search for uORFs. The second step is to perform homology searches of uORF amino acid sequences against EST databases using tBLASTn. The third step is the selection of uORFs on the basis of conservation of the stop codon position. The fourth step is the selection of uORFs conserved across a wide range of species by extracting uORFs whose tBLASTn hit-ESTs with conserved stop codons are found in each of multiple taxonomic categories. The fifth and sixth steps are filtering processes that exclude ‘spurious’ conserved uORFs. In the fifth step, we remove uORFs with sequences that are contained within an mORF of a separate transcript, such as a splice variant or a transcript from an overlapping gene. In the sixth step, we select uORFs conserved among homologous genes from diverse species to exclude spurious conserved uORFs that were extracted based on false-positive BLAST hits to biologically unrelated genes.
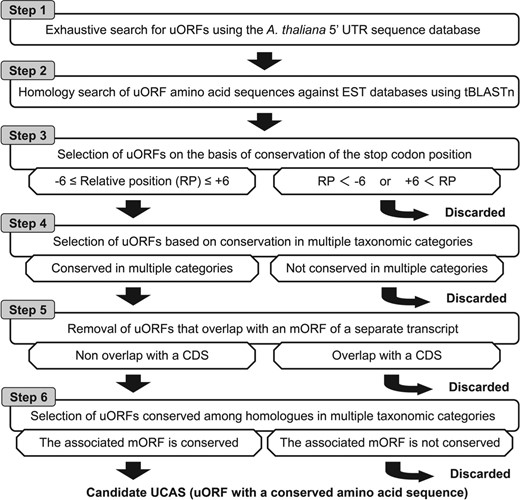
Outline of genome-wide search for A. thaliana UCASs using BAIUCAS
2.2 First step of BAIUCAS: exhaustive extraction of uORFs
In the first step of BAIUCAS, we identified uORFs by searching for sets of start and stop codons in a 5′-UTR sequence database. In this study, we used the TAIR10 A. thaliana 5′-UTR sequence database available on the Arabidopsis Information Resource (TAIR) website (http://arabidopsis.org/).Although uORFs that overlap their associated mORF are usually considered uORFs, we focused on the type of uORF that has both the start and stop codons within the 5′-UTR. In this study, we extracted each ORF that began with an ATG codon and ended with a stop codon as a different uORF, even if some uORFs share the same stop codon.
2.3 Second step of BAIUCAS: homology search of uORF sequences using BLAST
In the second step, the nucleotide sequences of the uORFs were translated into amino acid sequences, and homology searches of uORF amino acid sequences were performed using tBLASTn (a BLAST program) against the EST datasets available in the NCBI database (http://www.ncbi.nlm.nih.gov).In this study, uORFs with amino acid sequences longer than five residues were used as the query sequences for homology searches, because peptides of less than six residues are too short to detect significant homology and the known shortest bioactive uORF peptide involved in translational control is six residues in length (Ruan et al., 1996).
2.4 Third step of BAIUCAS: selection algorithm based on conservation of the stop codon position
In the third step of BAIUCAS, uORFs with conserved stop codon positions were selected since the stop codon positions are well conserved in previously documented sequence-dependent regulatory uORFs (Franceschetti et al., 2001; Ivanov et al., 2008; Jousse et al., 2001; Parola and Kobilka, 1994; Spevak et al., 2010; Wiese et al., 2004). For this purpose, we developed an algorithm to calculate the relative position of the stop codons between the uORFs and their tBLASTn-hit EST sequences as follows (Fig. 2). First, the entire nucleotide sequences of the tBLASTn-hit ESTs were obtained from the GenBank EST database, and then translated into amino acid sequences to determine the position of the stop codon. Next, the relative position of the stop codon between the uORF and the translated EST sequence was calculated based on the distance between each stop codon and the C-terminal amino acid residue in the sequence alignment of the tBLASTn results.
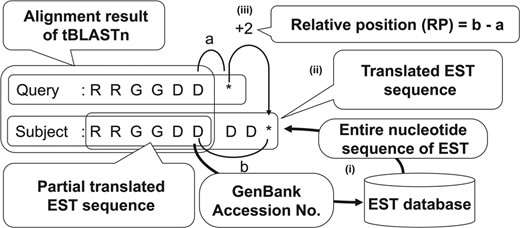
Calculation algorithm for the relative position of uORF stop codons in the third step of BAIUCAS. The illustration depicts the process to determine the relative position of the stop codons in the uORF sequences and tBLASTn-hit EST sequences. (i) Because only partial translated EST sequences are shown in the alignments of the tBLASTn search in Step 2, entire EST sequences were obtained from the GenBank EST database to determine the position of the stop codon in each EST. (ii) The entire nucleotide sequences of the ESTs were translated into amino acid sequences. (iii) The relative positions of the stop codons in the uORFs and the translated EST sequences were calculated based on the distances between each stop codon and the C-terminal amino acid residue in the sequence alignments of the tBLASTn results. The asterisks indicate stop codons
2.5 Fourth step of BAIUCAS: selection algorithm based on conservation across taxonomic categories
Among closely related species, uORF sequences can be retained by chance alone, independent of the function of the peptides they encode (Neafsey and Galagan, 2007). Therefore, as the fourth step of BAIUCAS, we devised an algorithm to find uORFs conserved across a wide range of species. In this algorithm, ESTs extracted in the third step are classified into multiple taxonomic categories according to the species from which the EST is derived, and uORFs with at least one EST in each of the taxonomic categories are selected (Fig. 3). In this study, to find A. thaliana uORFs conserved across eudicot plants, we divided eudicot species into the following three categories: (i) plants belonging to the order Brassicales, (ii) plants belonging to rosids excluding those belonging to the Brassicales and (iii) plants belonging to eudicots excluding those belonging to the rosids. To classify ESTs into these three categories, the genus names of the species of the ESTs were extracted using information in the GenBank EST database, as listed in Supplementary Table S1. Based on the genera, the ESTs were classified into the three aforementioned categories using the NCBI taxonomy database (http://www.ncbi.nlm.nih.gov/guide/taxonomy/).We tested two different selection criteria, Selection A and B. In Selection A, we selected uORFs that had at least one extracted EST in each of the three categories (Fig. 3A). uORFs conserved beyond the rosid group are expected to be extracted by this selection. Because the rosid group is one of two large groups in the eudicots, it is expected that selection of uORFs conserved beyond rosids will enable us to identify uORFs conserved across a wide range of eudicots. In Selection B, we selected uORFs that had at least one extracted EST in both of the categories (i) and (ii) (Fig. 3B). uORFs conserved beyond Brassicales are expected to be extracted by this selection. This selection will allow us to identify uORFs conserved among smaller taxonomic groups than Selection A.
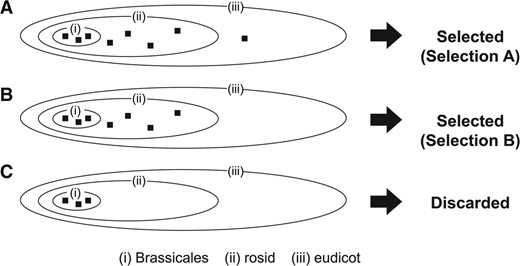
Selection of uORFs based on conservation across taxonomic categories in the fourth step of BAIUCAS. The ellipses (i), (ii) and (iii) represent the three eudicot categories defined in this study (see Methods). The filled squares in the ellipses show the tBLASTn-hit ESTs with conserved stop codons that were classified into the three categories. As illustrated in (A) and (B), we selected uORFs that had at least one tBLASTn hit in each of the three categories (Selection A) or each of the categories (i) and (ii) (Selection B). As illustrated in (C), we filtered out uORFs that had tBLASTn hits only in Brassicales
2.6 Fifth step of BAIUCAS: decision algorithm for the removal of uORFs that overlap with a CDS
In cases where a uORF is contained within the mORF of another splice variant (Fig. 4A), the uORF sequence may be conserved due to functional preservation of the protein encoded by the mORF, independent of the function of the uORF. In addition, in cases where the 5′-UTR of a gene overlaps with the mORF region of another gene, the uORF sequence in the 5′-UTR may be conserved due to functional preservation of the protein encoded by the overlapping mORF (Fig. 4B). To exclude these types of spurious conserved uORFs, as the fifth step of BAIUCAS, we developed an algorithm to examine whether uORFs overlap with mORFs of other transcripts by comparing the positions of the uORFs and mORFs in the genome. In this study, to determine the start and end positions of the uORFs in the A. thaliana genome, the positions of the start and end sites of the 5′-UTRs and the splice sites in the genome were obtained from the TAIR10 5′-UTR database, and the start and end positions of the uORFs in 5′-UTR sequences were calculated. The start and end positions of the mORFs in the A. thaliana genome were obtained from the TAIR10 CDS database. To determine whether the candidate uORFs overlap with the mORFs of other splice variants, the start and end positions of the uORFs were compared with those of the CDSs with the same AGI code (locus ID) as the 5′-UTR that contains the uORF. To investigate whether the candidate uORFs overlap with mORFs of other genes, the start and end positions of the uORFs were compared with those of CDSs with different AGI codes from the 5′-UTR that contains the uORF. In both cases, a uORF was filtered out if the start and/or end sites of the uORF were located between the start and end sites of a CDS.
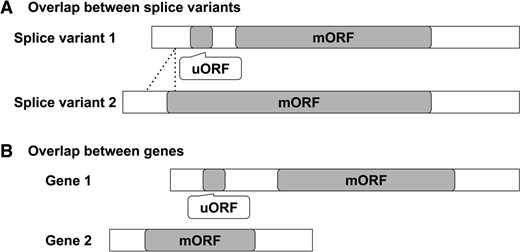
Decision algorithm for the elimination of uORFs that overlap with a CDS in the fifth step of BAIUCAS. The two types of spurious conserved uORFs removed in this step are shown: (A) uORFs with sequences that are contained within the mORF of another splice variant and (B) uORFs that overlap with the mORF of another gene
2.7 Sixth step of BAIUCAS: filtering algorithm based on homology between the mORF sequences
To exclude uORFs extracted based on false-positive BLAST hits to biologically unrelated genes, in the sixth step, we determined whether the candidate uORFs and their tBLASTn-hit ESTs were derived from homologous genes. For this purpose, we developed an algorithm to extract putative partial mORF sequences from the EST sequences and examine whether the amino acid sequence encoded by the putative partial mORF was similar to that of the mORF associated with the candidate uORF (Fig. 5). Putative partial mORFs were extracted from the EST sequences by searching for ATG codons located downstream of the conserved stop codons found in the third step. We defined the longest ORF as a putative mORF, irrespective of the presence or absence of an in-frame stop codon. The extracted putative mORF sequences were translated into amino acid sequences, and BLASTp searches were performed using the sequences as queries to evaluate whether they were similar to the protein encoded by the mORF associated with the corresponding candidate uORF. In this study, the minimum query sequence length was limited to 30 amino acids, and various E-value thresholds were tested ranging from 1e−1 to 1e−10. If the amino acid sequence of the putative mORF in an EST showed similarity to the A. thaliana protein derived from the same gene as the candidate uORF, we concluded that the uORF and the EST were derived from homologous genes. In this study, we selected uORFs with tBLASTn-hit ESTs that were confirmed to be derived from homologous genes in at least one species of each taxonomic category used in the fourth step.
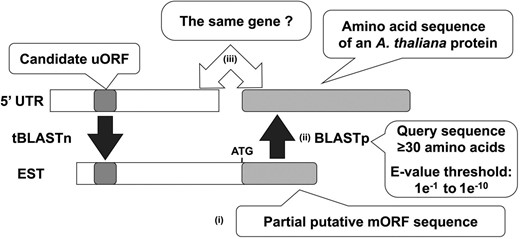
Selection algorithm for uORFs based on homology between the mORF sequences associated with the uORFs in the sixth step of BAIUCAS. The illustration depicts the process in the algorithm to determine whether a candidate uORF is conserved among homologous genes. (i) A putative partial mORF sequence was extracted from each tBLASTn-hit EST by searching for ATG codons located downstream of the conserved stop codon. We defined the longest ORF as the putative mORF, irrespective of the presence or absence of an in-frame stop codon. (ii) Using the amino acid sequences of the putative partial mORFs as query sequences, BLASTp searches were performed against the A. thaliana protein sequences in the UniProt database. (iii) If the amino acid sequence of the partial putative mORF shows similarity to an A. thaliana protein derived from the same gene as the candidate uORF, we concluded that the uORF and the EST were derived from homologous genes
3 RESULTS
3.1 Genome-wide search for A. thaliana UCASs using BAIUCAS/
Using BAIUCAS, which was developed in this study, we exhaustively searched for A. thaliana uORFs whose amino acid sequences are conserved across a wide range of eudicot plants. In the first step, 25 036 uORFs of 6960 genes were extracted from the 27 101 5′-UTR sequences of A. thaliana (Supplementary Table S2). In the second step, 16 913 uORF amino acid sequences, which were longer than five residues, were used as query sequences for tBLASTn searches. The tBLASTn searches were performed against the following NCBI EST datasets: est_crei, est_gmax, est_hvul, est_mtru, est_osat, est_slyc, est_taes, est_zmay and est_rest, such that all plant EST data available at NCBI were included. Arabidopsis ESTs were excluded from the tBLASTn searches. To identify short bioactive uORF peptides, the tBLASTn searches were conducted with an E-value cutoff of 1000. In total, 2 293 728 ESTs were detected by these searches, and 4220 uORFs of 1780 genes were extracted (Supplementary Table S2). To evaluate the performance of BAIUCAS, we checked how many CPUs, which are UCASs previously identified by Hayden and Jorgensen (2007), were extracted in each step of BAIUCAS. Although 54 of 58 CPUs were extracted in the second step, four CPUs were filtered out because no tBLASTn hit was found (Table 1). Most likely, this is due to a lack of ESTs containing these CPU sequences in the EST database.
Extraction of previously reported CPUs by BAIUCAS
| CPUs reported by Hayden and Jorgensen | BAIUCAS steps | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene (AGI code) | Homology group number | Step 1 | Step 2 | Step 3 | Step 4 | Three eudicot categories | Step 5 | Step6a | |||
| (i) | (ii) | (iii) | Splice variants | Other genes | |||||||
| (A) CPUs identified by Arabidopsis-rice comparison | |||||||||||
| AT1G75390 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G18160 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G62420 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G34590 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G49450 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT1G06150 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G27230 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G31280 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | NE | – | – |
| AT3G02470 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G15950 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G25570 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25670 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25690 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G52550 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G61230 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G07840 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G43020 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G59050 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G36730 | 7 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G12010 | 8 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09670 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G64550 | 9 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE e | – | – | – |
| AT1G64140 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G45430 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G19110 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G12430 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G22590 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G70780 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G23150 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G18000 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G48600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G73600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT3G01470 | 14 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G29950 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G50010 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT5G64340 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09460 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G51630 | 16 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G58120 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G53400 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G03190 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | NE | – | – |
| AT5G01710 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G36990 | 18 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G53590 | 19 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| (B) CPUs identified by Arabidopsis–Arabidopsis comparison | |||||||||||
| AT3G53670 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G37480 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G68550 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G25470 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G16860 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE d | – | – | – |
| AT1G78880 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G64630 | 23 | ✓ | ✓ | ✓ | ✓ f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT5G41990 | 23 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT3G22970 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT4G14620 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT3G45240 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G60550 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G10910 | 26 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT5G05280 | 26 | ✓ | NE | – | – | – | – | – | – | – | – |
| CPUs reported by Hayden and Jorgensen | BAIUCAS steps | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene (AGI code) | Homology group number | Step 1 | Step 2 | Step 3 | Step 4 | Three eudicot categories | Step 5 | Step6a | |||
| (i) | (ii) | (iii) | Splice variants | Other genes | |||||||
| (A) CPUs identified by Arabidopsis-rice comparison | |||||||||||
| AT1G75390 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G18160 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G62420 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G34590 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G49450 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT1G06150 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G27230 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G31280 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | NE | – | – |
| AT3G02470 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G15950 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G25570 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25670 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25690 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G52550 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G61230 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G07840 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G43020 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G59050 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G36730 | 7 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G12010 | 8 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09670 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G64550 | 9 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE e | – | – | – |
| AT1G64140 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G45430 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G19110 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G12430 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G22590 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G70780 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G23150 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G18000 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G48600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G73600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT3G01470 | 14 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G29950 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G50010 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT5G64340 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09460 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G51630 | 16 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G58120 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G53400 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G03190 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | NE | – | – |
| AT5G01710 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G36990 | 18 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G53590 | 19 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| (B) CPUs identified by Arabidopsis–Arabidopsis comparison | |||||||||||
| AT3G53670 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G37480 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G68550 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G25470 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G16860 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE d | – | – | – |
| AT1G78880 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G64630 | 23 | ✓ | ✓ | ✓ | ✓ f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT5G41990 | 23 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT3G22970 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT4G14620 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT3G45240 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G60550 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G10910 | 26 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT5G05280 | 26 | ✓ | NE | – | – | – | – | – | – | – | – |
NE, not extracted; (–), not analyzed
aSee Supplementary Tables S3 and S4 for E-value thresholds used for BLASTp to extract each CPUs.
bExtracted by Selection A and B.
cCPUs are registered in the TAIR CDS database as CDSs with an AGI code different from that of the associated mORF; therefore, the CPUs were removed as uORFs that overlap with a CDS.
dThe stop codon position of the uORF was not conserved.
eNo tBLASTn hit was found.
fExtracted by Selection B.
Extraction of previously reported CPUs by BAIUCAS
| CPUs reported by Hayden and Jorgensen | BAIUCAS steps | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene (AGI code) | Homology group number | Step 1 | Step 2 | Step 3 | Step 4 | Three eudicot categories | Step 5 | Step6a | |||
| (i) | (ii) | (iii) | Splice variants | Other genes | |||||||
| (A) CPUs identified by Arabidopsis-rice comparison | |||||||||||
| AT1G75390 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G18160 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G62420 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G34590 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G49450 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT1G06150 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G27230 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G31280 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | NE | – | – |
| AT3G02470 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G15950 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G25570 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25670 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25690 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G52550 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G61230 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G07840 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G43020 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G59050 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G36730 | 7 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G12010 | 8 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09670 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G64550 | 9 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE e | – | – | – |
| AT1G64140 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G45430 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G19110 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G12430 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G22590 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G70780 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G23150 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G18000 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G48600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G73600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT3G01470 | 14 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G29950 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G50010 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT5G64340 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09460 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G51630 | 16 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G58120 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G53400 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G03190 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | NE | – | – |
| AT5G01710 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G36990 | 18 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G53590 | 19 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| (B) CPUs identified by Arabidopsis–Arabidopsis comparison | |||||||||||
| AT3G53670 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G37480 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G68550 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G25470 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G16860 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE d | – | – | – |
| AT1G78880 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G64630 | 23 | ✓ | ✓ | ✓ | ✓ f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT5G41990 | 23 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT3G22970 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT4G14620 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT3G45240 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G60550 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G10910 | 26 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT5G05280 | 26 | ✓ | NE | – | – | – | – | – | – | – | – |
| CPUs reported by Hayden and Jorgensen | BAIUCAS steps | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene (AGI code) | Homology group number | Step 1 | Step 2 | Step 3 | Step 4 | Three eudicot categories | Step 5 | Step6a | |||
| (i) | (ii) | (iii) | Splice variants | Other genes | |||||||
| (A) CPUs identified by Arabidopsis-rice comparison | |||||||||||
| AT1G75390 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G18160 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G62420 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G34590 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G49450 | 1 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT1G06150 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G27230 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT2G31280 | 2 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | NE | – | – |
| AT3G02470 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G15950 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT3G25570 | 3 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25670 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT4G25690 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G52550 | 4 | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ | NEc | ✓ |
| AT5G61230 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G07840 | 5 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G43020 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G59050 | 6 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G36730 | 7 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G12010 | 8 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09670 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G64550 | 9 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE e | – | – | – |
| AT1G64140 | 9 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G45430 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G19110 | 10 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G12430 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G22590 | 11 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G70780 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G23150 | 12 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G18000 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G48600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT1G73600 | 13 | ✓ | NE | – | – | – | – | – | – | – | – |
| AT3G01470 | 14 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G29950 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G50010 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT5G64340 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G09460 | 15 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G51630 | 16 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G58120 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G53400 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G03190 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | NE | – | – |
| AT5G01710 | 17 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT4G36990 | 18 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G53590 | 19 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| (B) CPUs identified by Arabidopsis–Arabidopsis comparison | |||||||||||
| AT3G53670 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT2G37480 | 20 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G68550 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT1G25470 | 21 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT1G16860 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE d | NE d | – | – | – |
| AT1G78880 | 22 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT1G64630 | 23 | ✓ | ✓ | ✓ | ✓ f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT5G41990 | 23 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | NE |
| AT3G22970 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT4G14620 | 24 | ✓ | ✓ | ✓ | ✓f | ✓ | ✓ | NE e | ✓ | NE c | ✓ |
| AT3G45240 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT5G60550 | 25 | ✓ | ✓ | ✓ | ✓ b | ✓ | ✓ | ✓ | ✓ | NE c | ✓ |
| AT3G10910 | 26 | ✓ | ✓ | ✓ | NE | ✓ | NE e | NE e | – | – | – |
| AT5G05280 | 26 | ✓ | NE | – | – | – | – | – | – | – | – |
NE, not extracted; (–), not analyzed
aSee Supplementary Tables S3 and S4 for E-value thresholds used for BLASTp to extract each CPUs.
bExtracted by Selection A and B.
cCPUs are registered in the TAIR CDS database as CDSs with an AGI code different from that of the associated mORF; therefore, the CPUs were removed as uORFs that overlap with a CDS.
dThe stop codon position of the uORF was not conserved.
eNo tBLASTn hit was found.
fExtracted by Selection B.
In the third step, the relative position of the stop codons between the uORFs and their tBLASTn-hit EST sequences were determined (see Methods and Fig. 2). To determine how much difference in the relative stop codon position should be allowed, we used CPUs. The threshold of the distance between the stop codon positions was varied ranging from 0 to 100 codons, and ESTs were extracted for each CPUs with the various thresholds. Using the extracted ESTs, we examined how many CPUs were extracted by Selection A in the fourth step, in which we selected uORFs with any extracted EST in all of the three eudicot categories defined in this study (see Methods and Fig. 3A). When the threshold was raised to six, the number of extracted CPUs reached to the maximum (Supplementary Fig. S1). On the basis of this result, we decided to filter out ESTs in which the difference in the relative stop codon position between the EST and the corresponding uORF is larger than six codons. In this step, 298 565 ESTs were extracted in total, which corresponds to 3365 uORFs of 1421 A. thaliana genes (Supplementary Table S2).
In the fourth step, we tested two different selection criteria, Selection A and B, as mentioned in Methods. In Selection A, by selecting uORFs with at least one extracted EST in each of the three eudicot categories, 849 uORFs of 372 genes were extracted (Supplementary Table S2). In Selection B, by selecting uORFs with at least one extracted EST in both of the categories (i) and (ii), 1046 uORFs of 464 genes were extracted (Supplementary Table S2). In this step, 46 CPUs were extracted by Selection A, whereas 49 CPUs were extracted by Selection B (Table 1).
In the fifth step, uORFs that overlap with an mORF of a separate transcript were removed. First, we filtered out uORFs that were contained within the mORF of another splice variant (see Methods and Fig. 4A). In total, 441 uORFs of 202 genes were extracted in this filtering process (Supplementary Table S2). Two of CPUs, AT2G31280 and AT5G03190 uORFs, were removed due to the presence of splice variants in which the region corresponding to the CPU was within the mORF (Table 1). As for AT5G03190, the presence of a splice variant harboring a uORF-mORF fusion was discussed by Hayden and Jorgensen (2007). Next, we eliminated uORFs that were contained within an mORF of an overlapping gene (see Methods and Fig. 4B). In this process, 242 uORFs of 129 genes were extracted (Supplementary Table S2). All of the remaining CPUs were filtered out in this step (Table 1). This is because we used the TAIR10 CDS database as the A. thaliana mORF sequence dataset, and the previously identified conserved uORF sequences are registered as CDS sequences in this database.
Since tBLASTn hits with high E-values were included in the second step, the homology search results may contain many false-positive hits to biologically unrelated sequences. To exclude uORFs based on false-positive BLAST hits, we examined whether the candidate uORFs and their tBLASTn-hit ESTs were derived from homologous genes by comparing their associated mORF sequences using BLASTp (see Methods and Fig. 5). In the BLASTp analysis, various E-value thresholds were tested ranging from 1e−1 to 1e−10. We extracted uORFs with tBLASTn-hit ESTs that were confirmed to be derived from homologous genes in at least one species in each of the taxonomic categories used in the fourth step. In this final step, 74 uORFs of 35 genes were extracted (Supplementary Table S2). As mentioned in the Methods, in this study, overlapping uORFs that began with different initiation codons but ended with the same stop codon were counted as different uORFs. If such uORFs are considered the same uORF, each of the extracted 35 genes contains a single candidate UCAS. Hereafter, we describe such overlapping uORFs as the same uORF, and therefore 35 uORFs of 35 genes were extracted as candidate UCASs using BAIUCAS. Among the 35 candidate UCASs, 30 of them were extracted even when the BLASTp analysis was performed at 1e−10 of E-value cutoff. The remaining five candidate UCASs, AT1G61460, AT2G05410, AT2G27350, AT3G08730 and AT5G02480 were extracted when the E-value threshold was raised to 1e−2, 1e−5, 1e−3, 1e−5 and 1e−4, respectively (Supplementary Tables S5 and S6). Using full-length protein sequences or assembled EST sequences, we further confirmed these candidate UCASs are conserved among homologous genes from diverse species.
As mentioned earlier, most CPUs were filtered out in the fifth step. To examine how many CPUs would have been extracted by BAIUCAS if they had not been eliminated for the artificial reason, the 46 CPUs that were removed in the second part of the fifth step were applied in the sixth step of BAIUCAS. As shown in Table 1, 43 CPUs were extracted in this analysis.
3.2 Validation of the candidate UCASs
Although we excluded uORFs whose sequence is a part of an mORF in the fifth step using the A. thaliana CDS database, our filter could have failed to remove this type of spurious conserved uORF if the sequence information in the CDS is incomplete. To test for this possibility in the 35 candidate UCASs extracted by BAIUCAS, we manually examined the uORF sequences by BLASTx to determine whether they were similar to protein sequences in any plant species. In this analysis, the nucleotide sequences of the 35 uORFs were translated in all three reading frames, and the deduced amino acid sequences were used as query sequences for homology searches against the UniProt protein database (http://www.uniprot.org/).Of the 35 uORFs, there were 12 with hits in the N-terminal region of proteins encoded by the mORFs of homologous genes from A. thaliana or other plants, suggesting that the regions corresponding to these uORF sequences are part of the mORFs of homologous genes. Therefore, conservation of these uORF sequences is most likely due to functional preservation of the proteins encoded by the mORFs. We discarded these 12 uORFs as spurious conserved uORFs. For seven of the 12 discarded candidate UCASs, we found A. thaliana proteins in which the region corresponding to the uORF was fused to the mORF. Comparisons of the protein sequences and the corresponding full-length cDNA sequences (TAIR10) showed that four of them appear to be spurious uORFs that arose due to errors in the cDNA sequences. For three of the seven discarded candidate UCASs, the BLASTx-hit A. thaliana proteins appeared to be derived from splice variants whose corresponding mORF sequence did not exist in the TAIR10 CDS database.
Besides the discarded candidate UCASs, The uORF sequence of the AT2G11890 gene matched an A. thaliana protein, which is similar to mammalian and fungal CDC26, a subunit of the anaphase-promoting complex that is involved in cell cycle regulation. The AT2G11890 uORF encodes the entire A. thaliana CDC26-like protein, and the size of the AT2G11890 uORF-encoded protein resembles those of the mammalian and fungal CDC26 proteins. In addition, no uORF-mORF fusion protein was found in any plant species as far as we searched by BLASTp using the AT2G11890 mORF-encoded protein sequence as query. These facts suggest that the uORF and the mORF in AT2G11890 encode separate proteins, and therefore we did not exclude this uORF from the candidate UCASs. In the ESTs derived from angiosperms and gymnosperms, an ORF encoding a CDC26-like protein is associated with a partial ORF whose translated sequence is similar to the N-terminal region of the adenylate cyclase. This suggests that the same transcript encodes a CDC26-like protein and an adenylate cyclase in a wide range of higher plants.
We also examined other plant homologues of the proteins encoded by the mORFs associated with the other remaining 23 candidate UCASs by searching for homologues with BLASTp, and confirmed that no homologous protein contain the region corresponding to the uORFs. This result suggests that these candidate UCASs are not part of mORFs but are authentic uORFs. From these candidate UCASs, we further excluded AT3G08720 and AT3G08730 uORFs, because these genes are orthologous to the rice LOC_Os03g21620 gene, which contains a conserved uORF identified by Tran et al.(2008), and the reading frames of these A. thaliana uORFs are different from those of uORFs conserved in other plant orthologs (see Supplementary Fig. S2 for details). Among the remaining 21 candidate UCASs, 13 of them were extracted by both Selection A and B, whereas 8 of them were only extracted by Selection B (Table 2).
Candidate UCASs extracted by BAIUCAS
| Gene (AGI code) | Gene symbol | mORF description | uORF lengtha | Median pairwise Ka/Ks value | P-valued | q valuee | ||
|---|---|---|---|---|---|---|---|---|
| uORF | mORFb | Controlc | ||||||
| (A) Extracted by Selection A and B | ||||||||
| AT1G30270 | CIPK23 | CBL-interacting protein kinase | 19 | 0.0134 | 0.0871 | 0.870 | 4.27E-59 | 2.99E-58 |
| AT1G67480 | – | Galactose oxidase/kelch repeat superfamily protein | 72 | 0.198 | 0.132 | 0.833 | 2.45E-37 | 8.58E-37 |
| AT2G11890 | – | Adenylate cyclase | 65 | 0.271 | 0.155 | 0.884 | 5.81E-67 | 6.10E-66 |
| AT2G22500 | DIC1, UCP5 | Mitochondrial dicarboxylate carrier | 35 | 0.0992 | 0.0337 | 0.883 | 1.65E-89 | 3.47E-88 |
| AT2G42880 | ATMPK20 | MAP kinase | 37 | 0.0214 | 0.0878 | 0.826 | 1.93E-36 | 5.79E-36 |
| AT3G15430 | – | RCC1 family protein | 48 | 0.0901 | 0.128 | 0.854 | 2.07E-11 | 3.11E-11 |
| AT4G10170 | – | SNARE-like superfamily protein | 55 | 0.127 | 0.122 | 0.832 | 2.82E-15 | 4.94E-15 |
| AT4G12790 | – | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 36 | 0.0533 | 0.107 | 0.771 | 3.42E-22 | 6.53E-22 |
| AT4G30960 | CIPK6 | CBL-interacting protein kinase | 32 | 0.262 | 0.0567 | 0.903 | 1.25E-38 | 5.25E-38 |
| AT5G02480 | – | HSP20-like chaperones superfamily protein | 32 | 0.0906 | NCf | 0.849 | 3.17E-05 | 3.70E-05 |
| AT5G09330 | ANAC082, VNI1 | NAC domain containing transcription factor | 37 | 0.160 | 0.202 | 0.845 | 2.68E-34 | 6.84E-34 |
| AT5G46590 | ANAC096 | NAC domain containing transcription factor | 40 | 0.0545 | 0.130 | 0.829 | 7.08E-23 | 1.49E-22 |
| AT5G64060 | ANAC103 | NAC domain containing transcription factor | 37 | 0.172 | 0.176 | 0.844 | 2.93E-34 | 6.84E-34 |
| (B) Extracted by Selection B | ||||||||
| AT1G55760 | – | BTB/POZ domain-containing protein | 26 | 0.732 | 0.133 | 0.850 | 0.354 | 0.354 |
| AT1G72510 | – | Protein of unknown function | 24 | 0.280 | 0.0800 | 0.800 | 0.0573 | 0.0633 |
| AT2G27350 | OTLD1 | Otubain-like deubiquitinase | 43 | 0.205 | 0.160 | 0.866 | 5.77E-43 | 3.03E-42 |
| AT3G49430 | AT-SR34A | Serine/arginine-rich proteinsplicing factor | 30 | 0.623 | 0.0909 | 0.775 | 0.171 | 0.180 |
| AT3G55050 | – | Protein phosphatase 2C family protein | 49 | 0.225 | 0.107 | 0.814 | 3.30E-13 | 5.33E-13 |
| AT5G27920 | – | F-box family protein | 34 | 0.161 | NCf | 0.851 | 5.53E-11 | 7.26E-11 |
| AT5G60450 | ARF4 | ARF family transcription factor | 27 | 0.300 | NCf | 0.911 | 9.24E-11 | 1.14E-10 |
| AT5G63640 | – | ENTH/VHS/GAT family protein | 27 | 0.0672 | NCf | 0.856 | 4.15E-11 | 5.81E-11 |
| Gene (AGI code) | Gene symbol | mORF description | uORF lengtha | Median pairwise Ka/Ks value | P-valued | q valuee | ||
|---|---|---|---|---|---|---|---|---|
| uORF | mORFb | Controlc | ||||||
| (A) Extracted by Selection A and B | ||||||||
| AT1G30270 | CIPK23 | CBL-interacting protein kinase | 19 | 0.0134 | 0.0871 | 0.870 | 4.27E-59 | 2.99E-58 |
| AT1G67480 | – | Galactose oxidase/kelch repeat superfamily protein | 72 | 0.198 | 0.132 | 0.833 | 2.45E-37 | 8.58E-37 |
| AT2G11890 | – | Adenylate cyclase | 65 | 0.271 | 0.155 | 0.884 | 5.81E-67 | 6.10E-66 |
| AT2G22500 | DIC1, UCP5 | Mitochondrial dicarboxylate carrier | 35 | 0.0992 | 0.0337 | 0.883 | 1.65E-89 | 3.47E-88 |
| AT2G42880 | ATMPK20 | MAP kinase | 37 | 0.0214 | 0.0878 | 0.826 | 1.93E-36 | 5.79E-36 |
| AT3G15430 | – | RCC1 family protein | 48 | 0.0901 | 0.128 | 0.854 | 2.07E-11 | 3.11E-11 |
| AT4G10170 | – | SNARE-like superfamily protein | 55 | 0.127 | 0.122 | 0.832 | 2.82E-15 | 4.94E-15 |
| AT4G12790 | – | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 36 | 0.0533 | 0.107 | 0.771 | 3.42E-22 | 6.53E-22 |
| AT4G30960 | CIPK6 | CBL-interacting protein kinase | 32 | 0.262 | 0.0567 | 0.903 | 1.25E-38 | 5.25E-38 |
| AT5G02480 | – | HSP20-like chaperones superfamily protein | 32 | 0.0906 | NCf | 0.849 | 3.17E-05 | 3.70E-05 |
| AT5G09330 | ANAC082, VNI1 | NAC domain containing transcription factor | 37 | 0.160 | 0.202 | 0.845 | 2.68E-34 | 6.84E-34 |
| AT5G46590 | ANAC096 | NAC domain containing transcription factor | 40 | 0.0545 | 0.130 | 0.829 | 7.08E-23 | 1.49E-22 |
| AT5G64060 | ANAC103 | NAC domain containing transcription factor | 37 | 0.172 | 0.176 | 0.844 | 2.93E-34 | 6.84E-34 |
| (B) Extracted by Selection B | ||||||||
| AT1G55760 | – | BTB/POZ domain-containing protein | 26 | 0.732 | 0.133 | 0.850 | 0.354 | 0.354 |
| AT1G72510 | – | Protein of unknown function | 24 | 0.280 | 0.0800 | 0.800 | 0.0573 | 0.0633 |
| AT2G27350 | OTLD1 | Otubain-like deubiquitinase | 43 | 0.205 | 0.160 | 0.866 | 5.77E-43 | 3.03E-42 |
| AT3G49430 | AT-SR34A | Serine/arginine-rich proteinsplicing factor | 30 | 0.623 | 0.0909 | 0.775 | 0.171 | 0.180 |
| AT3G55050 | – | Protein phosphatase 2C family protein | 49 | 0.225 | 0.107 | 0.814 | 3.30E-13 | 5.33E-13 |
| AT5G27920 | – | F-box family protein | 34 | 0.161 | NCf | 0.851 | 5.53E-11 | 7.26E-11 |
| AT5G60450 | ARF4 | ARF family transcription factor | 27 | 0.300 | NCf | 0.911 | 9.24E-11 | 1.14E-10 |
| AT5G63640 | – | ENTH/VHS/GAT family protein | 27 | 0.0672 | NCf | 0.856 | 4.15E-11 | 5.81E-11 |
a The length of the amino acid sequences of the candidate UCASs.
b The Ka/Ks values for mORFs associated with the uORFs were calculated by using only mORF sequences from species in which full-length mORF sequence information is available (see Supplementary Table S7).
c Negative control median Ka/Ks values calculated based on the distribution of Ka/Ks values for artificially mutated uORF sequences (see Supplementary Text 1 for details).
d P-values calculated by the U-test for the distribution of Ka/Ks values of uORFs and negative controls.
e Adjustment for multiple comparisons was conducted by controlling the false discovery rate using Benjamini and Hochberg (1995) procedure.
f NC, not calculated, because full-length sequence information of the associated mORFs is unavailable besides A. thaliana.
Candidate UCASs extracted by BAIUCAS
| Gene (AGI code) | Gene symbol | mORF description | uORF lengtha | Median pairwise Ka/Ks value | P-valued | q valuee | ||
|---|---|---|---|---|---|---|---|---|
| uORF | mORFb | Controlc | ||||||
| (A) Extracted by Selection A and B | ||||||||
| AT1G30270 | CIPK23 | CBL-interacting protein kinase | 19 | 0.0134 | 0.0871 | 0.870 | 4.27E-59 | 2.99E-58 |
| AT1G67480 | – | Galactose oxidase/kelch repeat superfamily protein | 72 | 0.198 | 0.132 | 0.833 | 2.45E-37 | 8.58E-37 |
| AT2G11890 | – | Adenylate cyclase | 65 | 0.271 | 0.155 | 0.884 | 5.81E-67 | 6.10E-66 |
| AT2G22500 | DIC1, UCP5 | Mitochondrial dicarboxylate carrier | 35 | 0.0992 | 0.0337 | 0.883 | 1.65E-89 | 3.47E-88 |
| AT2G42880 | ATMPK20 | MAP kinase | 37 | 0.0214 | 0.0878 | 0.826 | 1.93E-36 | 5.79E-36 |
| AT3G15430 | – | RCC1 family protein | 48 | 0.0901 | 0.128 | 0.854 | 2.07E-11 | 3.11E-11 |
| AT4G10170 | – | SNARE-like superfamily protein | 55 | 0.127 | 0.122 | 0.832 | 2.82E-15 | 4.94E-15 |
| AT4G12790 | – | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 36 | 0.0533 | 0.107 | 0.771 | 3.42E-22 | 6.53E-22 |
| AT4G30960 | CIPK6 | CBL-interacting protein kinase | 32 | 0.262 | 0.0567 | 0.903 | 1.25E-38 | 5.25E-38 |
| AT5G02480 | – | HSP20-like chaperones superfamily protein | 32 | 0.0906 | NCf | 0.849 | 3.17E-05 | 3.70E-05 |
| AT5G09330 | ANAC082, VNI1 | NAC domain containing transcription factor | 37 | 0.160 | 0.202 | 0.845 | 2.68E-34 | 6.84E-34 |
| AT5G46590 | ANAC096 | NAC domain containing transcription factor | 40 | 0.0545 | 0.130 | 0.829 | 7.08E-23 | 1.49E-22 |
| AT5G64060 | ANAC103 | NAC domain containing transcription factor | 37 | 0.172 | 0.176 | 0.844 | 2.93E-34 | 6.84E-34 |
| (B) Extracted by Selection B | ||||||||
| AT1G55760 | – | BTB/POZ domain-containing protein | 26 | 0.732 | 0.133 | 0.850 | 0.354 | 0.354 |
| AT1G72510 | – | Protein of unknown function | 24 | 0.280 | 0.0800 | 0.800 | 0.0573 | 0.0633 |
| AT2G27350 | OTLD1 | Otubain-like deubiquitinase | 43 | 0.205 | 0.160 | 0.866 | 5.77E-43 | 3.03E-42 |
| AT3G49430 | AT-SR34A | Serine/arginine-rich proteinsplicing factor | 30 | 0.623 | 0.0909 | 0.775 | 0.171 | 0.180 |
| AT3G55050 | – | Protein phosphatase 2C family protein | 49 | 0.225 | 0.107 | 0.814 | 3.30E-13 | 5.33E-13 |
| AT5G27920 | – | F-box family protein | 34 | 0.161 | NCf | 0.851 | 5.53E-11 | 7.26E-11 |
| AT5G60450 | ARF4 | ARF family transcription factor | 27 | 0.300 | NCf | 0.911 | 9.24E-11 | 1.14E-10 |
| AT5G63640 | – | ENTH/VHS/GAT family protein | 27 | 0.0672 | NCf | 0.856 | 4.15E-11 | 5.81E-11 |
| Gene (AGI code) | Gene symbol | mORF description | uORF lengtha | Median pairwise Ka/Ks value | P-valued | q valuee | ||
|---|---|---|---|---|---|---|---|---|
| uORF | mORFb | Controlc | ||||||
| (A) Extracted by Selection A and B | ||||||||
| AT1G30270 | CIPK23 | CBL-interacting protein kinase | 19 | 0.0134 | 0.0871 | 0.870 | 4.27E-59 | 2.99E-58 |
| AT1G67480 | – | Galactose oxidase/kelch repeat superfamily protein | 72 | 0.198 | 0.132 | 0.833 | 2.45E-37 | 8.58E-37 |
| AT2G11890 | – | Adenylate cyclase | 65 | 0.271 | 0.155 | 0.884 | 5.81E-67 | 6.10E-66 |
| AT2G22500 | DIC1, UCP5 | Mitochondrial dicarboxylate carrier | 35 | 0.0992 | 0.0337 | 0.883 | 1.65E-89 | 3.47E-88 |
| AT2G42880 | ATMPK20 | MAP kinase | 37 | 0.0214 | 0.0878 | 0.826 | 1.93E-36 | 5.79E-36 |
| AT3G15430 | – | RCC1 family protein | 48 | 0.0901 | 0.128 | 0.854 | 2.07E-11 | 3.11E-11 |
| AT4G10170 | – | SNARE-like superfamily protein | 55 | 0.127 | 0.122 | 0.832 | 2.82E-15 | 4.94E-15 |
| AT4G12790 | – | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 36 | 0.0533 | 0.107 | 0.771 | 3.42E-22 | 6.53E-22 |
| AT4G30960 | CIPK6 | CBL-interacting protein kinase | 32 | 0.262 | 0.0567 | 0.903 | 1.25E-38 | 5.25E-38 |
| AT5G02480 | – | HSP20-like chaperones superfamily protein | 32 | 0.0906 | NCf | 0.849 | 3.17E-05 | 3.70E-05 |
| AT5G09330 | ANAC082, VNI1 | NAC domain containing transcription factor | 37 | 0.160 | 0.202 | 0.845 | 2.68E-34 | 6.84E-34 |
| AT5G46590 | ANAC096 | NAC domain containing transcription factor | 40 | 0.0545 | 0.130 | 0.829 | 7.08E-23 | 1.49E-22 |
| AT5G64060 | ANAC103 | NAC domain containing transcription factor | 37 | 0.172 | 0.176 | 0.844 | 2.93E-34 | 6.84E-34 |
| (B) Extracted by Selection B | ||||||||
| AT1G55760 | – | BTB/POZ domain-containing protein | 26 | 0.732 | 0.133 | 0.850 | 0.354 | 0.354 |
| AT1G72510 | – | Protein of unknown function | 24 | 0.280 | 0.0800 | 0.800 | 0.0573 | 0.0633 |
| AT2G27350 | OTLD1 | Otubain-like deubiquitinase | 43 | 0.205 | 0.160 | 0.866 | 5.77E-43 | 3.03E-42 |
| AT3G49430 | AT-SR34A | Serine/arginine-rich proteinsplicing factor | 30 | 0.623 | 0.0909 | 0.775 | 0.171 | 0.180 |
| AT3G55050 | – | Protein phosphatase 2C family protein | 49 | 0.225 | 0.107 | 0.814 | 3.30E-13 | 5.33E-13 |
| AT5G27920 | – | F-box family protein | 34 | 0.161 | NCf | 0.851 | 5.53E-11 | 7.26E-11 |
| AT5G60450 | ARF4 | ARF family transcription factor | 27 | 0.300 | NCf | 0.911 | 9.24E-11 | 1.14E-10 |
| AT5G63640 | – | ENTH/VHS/GAT family protein | 27 | 0.0672 | NCf | 0.856 | 4.15E-11 | 5.81E-11 |
a The length of the amino acid sequences of the candidate UCASs.
b The Ka/Ks values for mORFs associated with the uORFs were calculated by using only mORF sequences from species in which full-length mORF sequence information is available (see Supplementary Table S7).
c Negative control median Ka/Ks values calculated based on the distribution of Ka/Ks values for artificially mutated uORF sequences (see Supplementary Text 1 for details).
d P-values calculated by the U-test for the distribution of Ka/Ks values of uORFs and negative controls.
e Adjustment for multiple comparisons was conducted by controlling the false discovery rate using Benjamini and Hochberg (1995) procedure.
f NC, not calculated, because full-length sequence information of the associated mORFs is unavailable besides A. thaliana.
To assess whether the sequences of the remaining 21 candidate UCASs are conserved at the nucleotide level or amino acid level, we calculated a ratio of non-synonymous to synonymous nucleotide substitutions (Ka/Ks) for each uORF (see Supplementary Text 1 for details of the method of the Ka/Ks analysis). A Ka/Ks ratio close to 1 indicates neutral evolution, whereas a Ka/Ks ratio close to 0 suggests that purifying selection acted on the amino acid sequences. Among the candidate UCASs, the Ka/Ks ratios of AT1G55760 and AT3G49430 uORFs, which were only extracted from Selection B, were close to those of negative controls (Table 2). Although the Ka/Ks ratio of the AT1G72510 uORF was as low as those of the other remaining candidate UCASs, it is not significantly different from that of its negative control. Therefore, we could not rule out the possibility that this uORF sequence is conserved at the nucleotide level. The Ka/Ks ratios of the other 18 candidate UCASs were equal to or less than 0.300, and significantly different from those of the negative controls (Table 2). This result suggests that these 18 uORF sequences are conserved at the amino acid level but not at the nucleotide level. On the basis of these results, we concluded that these 18 uORFs are UCASs.
Among the 18 UCAS-containing genes, AT5G09330 and AT5G64060 are paralogous genes and their uORF sequences are similar to each other. Besides these, nine of the 18 UCAS-containing genes have paralogous genes. However, in three of the nine genes, their paralogues lack 5′-UTR sequence information, and therefore we could not address whether the UCAS sequences are conserved in the paralogues. As for the remaining six genes, 5′-UTR sequence information of their paralogues is available. However, none of the 5′-UTR sequences shows similarity to the corresponding UCAS sequence, suggesting that the UCAS sequences are not conserved in the paralogues.
Alignments of the 17 groups of UCASs and their homologous uORF sequences from other plants are shown in Fig. 6. In these alignments, we added uORF sequences derived from species in which the uORF-containing gene was confirmed to be homologous to the corresponding A. thaliana UCAS-containing gene by using assembled EST, cDNA or protein sequences. Eight of the 12 UCAS groups extracted from Selection A are conserved beyond dicots. In contrast, all of the five UCASs extracted only from Selection B are only conserved among dicots, and one of them are only conserved among rosids.


continued. Alignment of conserved uORF sequences identified in this study. The amino acid sequences of the uORFs were aligned using ClustalW ver. 1.83 and displayed using Jalview ver. 2.7. All sequences shown are the entire translated uORF sequences. Most of the sequences are supported by at least two ESTs, although some sequences are supported by only one EST (see Supplementary Data). aAquilegia formosa × Aquilegia pubescens. bMalus × domestica × Malus sieversii. cPopulus trichocarpa × Populus deltoides
4 DISCUSSION
In this study, we developed BAIUCAS, a novel algorithm to identify UCASs and identified 18 novel UCASs in A. thaliana using BAIUCAS. The main feature of BAIUCAS is the comparison of uORF sequences with EST sequences from a large number of species and the selection of uORFs conserved across multiple taxonomic categories. This feature increases the chances of identifying UCASs compared with conventional comparative genomic approaches in which the uORF sequences are compared between a few selected species, because BAIUCAS can identify a UCAS even if it is not conserved in certain species. In fact, conserved uORFs corresponding to some of the UCASs were not found in certain taxonomic groups. For example, although the AT4G12790 UCAS is conserved in dicots and monocots, corresponding conserved uORFs were only found in Solanales among asterids, which is one of two large eudicot groups. In addition, although the AT1G30270 UCAS is conserved in angiosperms and gymnosperms, no corresponding conserved uORF was found in homologous genes of monocots. This feature of BAIUCAS is considered particularly advantageous for the identification of conserved uORFs with only a small number of conserved amino acid residues, because detection of the homology of such uORFs is largely dependent on the selection of species for comparison. In fact, we identified UCASs, such as AT1G30270 and AT3G55050, with relatively short conserved sequences. In addition, we identified UCASs, such as AT2G42880 and AT5G46590, with relatively low sequence conservation in A. thaliana and only a small number of conserved amino acid residues (Fig. 6). In this study, we tested two selection criteria in the fourth step of BAIUCAS, and demonstrated that, by using different selection criteria regarding taxonomic categories, BAIUCAS can extract uORFs conserved among different ranges of taxonomic groups. Although the selection of uORFs conserved among smaller taxonomic groups [i.e. categories (i) and (ii)] increased the risk of extracting uORFs whose sequences are retained at the nucleotide level, such a selection led us to identify UCASs more comprehensively.
The UCASs identified in this study can be classified into the following two groups according to sequence conservation patterns: (I) uORFs with highly conserved C-terminal regions and perfectly conserved stop codon positions and (II) uORFs in which the entire sequences or the N-terminal and middle regions are conserved. Group I is composed of 10 groups of UCASs (Fig. 6, F, G, H, L, K, M, N, O and P), whereas Group II consists of seven UCASs (Fig. 6A, C, D, E, I, J and Q). In contrast to the Group I UCASs, the position of the stop codons is not perfectly conserved in the Group II UCASs, and differences in several codons of the stop codon positions are observed among species. The features of the Group I UCASs are consistent with those of previously reported sequence-dependent regulatory uORFs, in some of which the importance of the C-terminal amino acid sequence for translational regulation has been experimentally demonstrated (Alderete et al., 1999; Spevak et al., 2010). For two of these regulatory uORFs, cryo-electron microscopy analyses have shown that the C-terminal regions of the uORF-encoded nascent peptides interact with the ribosomal exit tunnel when ribosome stalling occurs (Bhushan et al., 2010). To assess whether the amino acid sequences of the UCASs identified in this study indeed have a regulatory function, experimental validation is necessary. For example, this can be validated by comparing the effects of amino acid sequence alterations and synonymous codon changes of uORFs on translation of the mORFs.
In addition to the identification of the 18 UCASs, we also demonstrated that BAIUCAS could extract 43 of the previously identified CPUs. Taking these CPUs into account, BAIUCAS could extract 61 A. thaliana UCASs, which are classified into 38 homology groups. Although BAIUCAS failed to extract 15 of the 58 CPUs, the reason why five of these were not extracted is that they did not fit one of our conditions that the amino acid sequence and the stop codon position of a uORF must be conserved in the multiple eudicot categories. Additionally, two of the CPUs were eliminated because of the condition that a uORF must not be within an mORF of a splice variant. In the sixth step, two of the CPUs were not extracted because of low conservation in the N-terminal region of the mORF-encoded protein. In this study, to assess whether A. thaliana uORFs and their tBLASTn-hit ESTs are derived from homologous genes, we directly compared A. thaliana protein sequences and amino acid sequences encoded by putative partial mORFs within the EST sequences. If the protein database expands to include a greater variety of plant species in the future, an alternative approach would be possible, in which full-length protein sequences corresponding to partial mORFs within the ESTs are compared with A. thaliana protein sequences. Such an approach may increase the number of conserved uORFs that are identifiable by BAIUCAS.
In conclusion, this study demonstrated that BAIUCAS is a powerful method for the identification of UCASs that makes it possible to more comprehensively identify conserved uORFs than conventional comparative genomic approaches. Because BAIUCAS is particularly useful for the identification of conserved uORFs with a small number of conserved amino acid residues, the algorithms used in BAIUCAS can be applied to the identification of short bioactive peptides besides uORF-encoded peptides, such as peptide hormones.
Funding: Grants-in-Aid for Scientific Research on Priority Areas (21027001 to H.O.) and for Young Scientists (B) (21710211 and 24710222 to H.T.) from the Ministry of Education, Science, Culture, and Sport of Japan, and the ‘Academic Frontier’ Project for Private Universities (matching fund subsidy from the Ministry of Education, Culture, Sports, Science and Technology [MEXT], 2005–2009).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alfonso Valencia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}