





Abstract
Summary: High-throughput sequencing has become an essential experimental approach for the investigation of transcriptional mechanisms. For some applications like ChIP-seq, several approaches for the prediction of peak locations exist. However, these methods are not designed for the identification of transcription start sites (TSSs) because such datasets contain qualitatively different noise.
In this application note, the R package TSSi is presented which provides a heuristic framework for the identification of TSSs based on 5′ mRNA tag data. Probabilistic assumptions for the distribution of the data, i.e. for the observed positions of the mapped reads, as well as for systematic errors, i.e. for reads which map closely but not exactly to a real TSS, are made and can be adapted by the user. The framework also comprises a regularization procedure which can be applied as a preprocessing step to decrease the noise and thereby reduce the number of false predictions.
Availability: The R package TSSi is available from the Bioconductor web site: www.bioconductor.org/packages/release/bioc/html/TSSi.html.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
High-throughput sequencing has become an essential experimental approach to investigate genomes and transcriptional processes. The core of most applications is a peak finding algorithm which is required to identify regions of interest. These algorithms are designed application specifically because the requirements depend on characteristics of the measurements as well as on the questions of interest. While cDNA sequencing (RNA-seq) using random priming and/or fragmentation of cDNA will result in a shallow distribution of reads typically biased toward the 3′ end, approaches like CAP-capture enrich 5′ ends of mRNAs and result in distinguishable peaks around the transcription start site(s) (TSSs). Similar methods can also be applied to specifically target 3′ ends of mRNAs. The resolution and utility of the multitude of end-tagging methods have been tremendously increased in recent years by the application of next-generation sequencing technologies which allow 5′ or 3′ digital gene expression at genome scale (Hoskins et al., 2011). When applied to sequencing of DNA fragments isolated by immunoprecipitation (ChIP-seq) broad and almost unimodal densities without gaps are obtained around a target position which, depending on the protein used for immunoprecipitation, usually represents a transcription factor binding site or provides insight into chromatin structure. For such applications, several approaches have been proposed although benchmark problems show that these algorithms still work insufficiently in some cases (references are provided in the Supplementary Material).
Predicting the location of TSSs is complicated by the possible existence of an unknown number of multiple, alternative TSSs. Furthermore, the transcription of many genes in eukaryotic genomes is initiated at not well-defined sharp or peaked sites, but in more fuzzily defined, broad transcription start regions (see references in the Supplementary Material). In addition, as illustrated in Figure 1, the measurements typically contain background reads, i.e. false positives not originating from real TSSs. Therefore, only the counts which are significantly larger than an expected number of background reads are intended to be predicted as TSSs. The 5′ end tag data used as a test set for our study (see Section 2) comprises reads mapping to 211 669 genomic positions. This vastly exceeds the expected number of TSSs, indicating the existence of reads not originating from real TSSs. Such reads cluster to certain genomic positions, i.e. the level of background noise seems to be proportional to proximate measurements yielding false positive reads preferably in regions of transcriptional activity. It is yet unclear, whether these are artifacts introduced from the experimental procedure or whether there is a biological meaning to this noise. As currently there is no error model available describing such noise, an heuristic approach is introduced for an automated and flexible prediction of TSSs in the following.
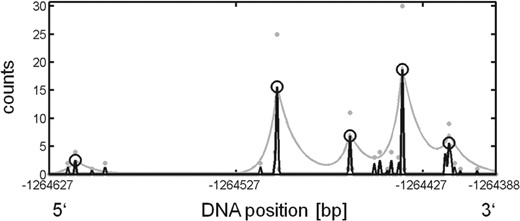
An example illustrating the results of the TSSi analysis for 5′ tag data. The horizontal axis is the genomic position of the transcribed DNA sequence, in this example on scaffold 1. Negative numbers on this axis indicate the anti-sense strand, i.e. transcription in the plot occurs from left to right. The raw reads at positions of the respective 5′ ends of the template DNA sequences are plotted as gray dots. The measurements processed by the regularization procedure are plotted as black lines and the predicted TSSs are depicted as black circles. The gray line indicates the contamination threshold level. For this illustration, λ1=1, λ2=0.1, τ5′=5 and τ3′=10 have been chosen
2 METHODOLOGY
As input, our method uses the number yi of sequenced transcripts as well as the genomic position i of the 5′ end of the mapped DNA sequences given by the chromosome/scaffold, the strand and the genomic position. As an example, we have analyzed 76 nt Illumina CAP-capture (5′ tag) data generated from protonema tissue of the moss Physcomitrella patens following the protocol of Maruyama and Sugano (1994). As a preprocessing of such data for use with TSSi reads need to be mapped to the reference genome to acquire read counts and position information. For computational efficiency, the analysis is iteratively performed on all segments with transcriptional activity. Further preprocessing, e.g. restriction to upstream regions of annotated genes, is optional (see Supplementary Material for details).
The first step in our analysis is a preprocessing procedure which reduces the noise by shrinking the counts toward zero. This step is intended to eliminate false positive counts as well as making further analyses more robust by reducing the impact of large counts. Such a shrinkage or regularization procedure constitutes a well-established strategy in statistics to make predictions conservative, i.e. to reduce the number of false positive predictions (Tibshirani, 1996).
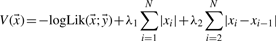
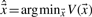 in a regularized manner. Here, N denotes the number of genomic positions, the vector
in a regularized manner. Here, N denotes the number of genomic positions, the vector  represents the true transcription level,
represents the true transcription level, 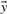 is the vector of measurements, i.e. the reads, and
is the vector of measurements, i.e. the reads, and 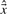 indicates the estimated level of transcription. The log-likelihood
indicates the estimated level of transcription. The log-likelihood 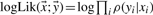 is given by the product of the probabilities of the counts yi which is assumed as a Poisson distribution
is given by the product of the probabilities of the counts yi which is assumed as a Poisson distribution 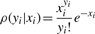
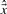 of the transcription levels
of the transcription levels  with a preferably small number of components, i.e. genomic positions, unequal to zero. The larger λ1, the more conservative is the identification procedure. To enhance the shrinkage of isolated counts in comparison to counts in regions of strong transcriptional activity, the information of consecutive genomic positions in the measurements is regarded by the third term in (1), i.e. by evaluating differences |xi−xi−1| between adjacent count estimates. Without regularization, i.e. for λ1=λ2=0,
with a preferably small number of components, i.e. genomic positions, unequal to zero. The larger λ1, the more conservative is the identification procedure. To enhance the shrinkage of isolated counts in comparison to counts in regions of strong transcriptional activity, the information of consecutive genomic positions in the measurements is regarded by the third term in (1), i.e. by evaluating differences |xi−xi−1| between adjacent count estimates. Without regularization, i.e. for λ1=λ2=0, 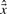 is the maximum-likelihood estimator of the expectation of the count distribution.
is the maximum-likelihood estimator of the expectation of the count distribution. is at least one read exceeding the expected number of false positive reads, i.e.
is at least one read exceeding the expected number of false positive reads, i.e. 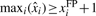 . The transcription levels
. The transcription levels 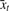 for all TSSs t are calculated by adding all counts xi to their nearest neighbor TSS. Then, the expected number of background reads is updated by convolution
for all TSSs t are calculated by adding all counts xi to their nearest neighbor TSS. Then, the expected number of background reads is updated by convolution 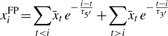
For the exemplary region displayed in Figure 1, the raw counts yi are plotted as gray dots, the regularized count estimates  are plotted as black lines, and the predicted TSSs are labeled by black circles. The exponentially decaying expected rates of background counts xFP are plotted as gray lines. In the Supplementary Material, the effects of changing the parameters λ1, λ2, τ5′ and τ3′ are illustrated.
are plotted as black lines, and the predicted TSSs are labeled by black circles. The exponentially decaying expected rates of background counts xFP are plotted as gray lines. In the Supplementary Material, the effects of changing the parameters λ1, λ2, τ5′ and τ3′ are illustrated.
3 CONCLUSIONS
An important application of high-throughput sequencing is the identification of TSSs. In this application note, the R package TSSi is introduced for the prediction of TSSs based on the mapped 5′ ends of mRNAs. As an optional preprocessing step, the transcription level is estimated by regularization to make the TSS predictions conservative.
Unless user-defined distributions are applied, a Poisson distribution for the counts and an exponentially decaying expected number of background reads proximate to TSSs are assumed. Since our implementation allows a flexible, project-specific adaptation, the method could also be valuable for peak finding problems in other applications.
The presented method does not use any prior information about potential TSSs, e.g. provided by annotation like transcription factor binding sites. Extending the method concerning such information could be a future task.
ACKNOWLEDGEMENTS
The authors acknowledge German Federal Ministry of Education and Research (BMBF) and German Research Foundation (DFG).
Funding: German Federal Ministry of Education and Research (BMBF) [0315766-VirtualLiver] and [0313921-FRISYS]; German Research Foundation (DFG) [RE 837/10-2].
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Ivo Hofacker


{kind=link}